Das MCAD-Hörbuch
Die Zip-Datei (596 MB) umfasst 45 MP3-Dateien, die alle Hauptartikel und die FAQs enthalten.








 Mein Name ist Magnus Rohleder und um es gleich mal vorwegzunehmen: Ich bin weder Arzt noch sonst wie medizinisch gebildeter Fachmann, sondern lediglich ein ehemals selbst vom MCAD-Mangel seines Kindes betroffener und an all diesen Zusammenhängen extrem interessierter Vater. Ich schreibe “ehemals”, weil sich dann herausgestellt hat, dass … aber der Reihe nach!
Mein Name ist Magnus Rohleder und um es gleich mal vorwegzunehmen: Ich bin weder Arzt noch sonst wie medizinisch gebildeter Fachmann, sondern lediglich ein ehemals selbst vom MCAD-Mangel seines Kindes betroffener und an all diesen Zusammenhängen extrem interessierter Vater. Ich schreibe “ehemals”, weil sich dann herausgestellt hat, dass … aber der Reihe nach!
Im Herbst 2007 sahen meine Frau und ich uns kurz nach der Geburt unseres Sohnes mit der Diagnose des uns völlig unbekannten MCAD-Mangels konfrontiert. Weder der sofort aufgesuchte Kinderarzt noch die Experten unserer Stoffwechselambulanz konnten uns auch nur ansatzweise zufriedenstellend über die Hintergründe dieser Stoffwechselstörung informieren und erst recht keine brauchbaren Ratschläge zum täglichen Umgang liefern. Da man uns seitens unserer Stoffwechselambulanz auch keine Kontakte zu anderen betroffenen Eltern vermitteln konnte, rief ich im Februar 2008 die Seite MCAD-Infos.de ins Leben, auf der ich die damals noch wenigen verfügbaren Informationen sammeln wollte.
Diese Fragen haben sich während der früheren Forumszeit und auch aus späteren Anfragen an mich als besonders dringlich herauskristallisiert und werden deshalb in diesem Abschnitt einzeln beantwortet.
Klicke dazu in der linken Spalte auf die dich interessierende Frage. Der zugehörige Artikel erscheint dann auf der rechten Seite, bzw. auf Handys unterhalb der kompletten Fragenliste.
Hätte ich diese ganzen Artikel und FAQs erst kürzlich erstellt, wäre es mir tatsächlich sehr wichtig gewesen, auf eine gendergerechte Schreibweise zu achten. In der Zeit, als ich die meisten der folgenden Inhalte geschrieben habe, war dies allerdings noch nirgends ein Thema. Jetzt beschäftigt mich der MCAD-Mangel jedoch nur noch nebenher, sodass es mir ehrlich gesagt ein zu großer Aufwand wäre, diese 210 Seiten Satz für Satz zu kontrollieren und umzuschreiben. Tatsächlich hatte ich damals beim Schreiben der Texte hauptsächlich Mütter als Leserinnen vor dem geistigen Auge, denn während der gesamten 10 Forumsjahre machten die sich registrierenden oder gar aktiv an den Diskussionen beteiligenden Väter betroffener Kinder nicht einmal 5% der Mitgliederzahl aus. Daher gehe ich einfach mal davon aus, dass es auch heute noch hauptsächlich die Mütter sind, die auf der Suche nach Informationen auf diese Seite stoßen und gerade diese Zeilen lesen. Aber wie auch immer, bitte störe dich nicht an meiner so gut wie nirgendwo gendergerechten Schreibweise und fühle dich bitte in jedem Fall angesprochen.
Und noch etwas: Auf dieser und weiteren Seiten erwähne ich vereinzelt, dass gewisse Aussagen seitens der (Stoffwechsel-)Kliniken vermutlich auf nicht allzu großer Erfahrung der euch behandelnden Ärzte mit dem MCAD-Mangel beruhen und daher teilweise an den Haaren herbeigezogen sind. Das darf man natürlich nicht verallgemeinern! Es gibt Stoffwechselkliniken mit einzelnen sich hervorragend mit dem MCAD-Mangel auskennenden Ärztinnen und Ärzten, von denen man wirklich bestens beraten wird. Wird man von diesen behandelt, bleibt kaum eine Frage offen! Die betreffenden Einrichtungen würde ich allerdings an einer einzigen Hand abzählen und dazu gehören auch nicht unbedingt die Kliniken mit den bekanntesten Namen. Immer noch sehr viel häufiger trifft man auch in den Stoffwechselambulanzen der Uni-Kliniken auf Ärzte, die von diesem speziellen Thema nur eine sehr vage Ahnung haben. Nur so lässt sich erklären, dass mir schon zu Forumszeiten und auch heute noch immer wieder von Eltern Fragen gestellt werden, die nur deshalb aufgekommen sind, weil ihnen gegenüber seitens ihrer manchmal auch noch häufig wechselnden Ärzte völlig haltlose und teilweise haarsträubende Aussagen gemacht wurden, die definitiv nicht auf großer Erfahrung, sondern auf weitestgehender Ahnungslosigkeit bzgl. des MCAD-Mangels beruhen. Um einige genau solcher Aussagen und deren Richtigstellung geht es in den FAQs und den weiteren Artikeln.![]()
Ja, die Chancen, dass es sich noch als falscher Alarm herausstellt, stehen zum jetzigen Zeitpunkt noch sehr gut, nämlich heutzutage bei etwa 52%. Soll heißen: für durchschnittlich etwa die Hälfte der Kinder, bei denen jedes Jahr das Neugeborenenscreening durch irgendwie auffällige Werte den Verdacht auf einen möglicherweise vorliegenden MCAD-Mangel aufkommen lässt, kann bereits nach dem anschließend durchzuführenden Kontrollscreening (“Konfirmations-Screening” oder “Recall”) Entwarnung gegeben werden, wenn die dann gemessenen Werte in den zugrunde gelegten Normbereichen liegen. Die konkreten Zahlen zeigt die folgende Tabelle, deren Daten den jährlichen Screeningreports der Deutschen Gesellschaft für Neugeborenenscreening entnommen sind:
In den letzten 4 aufgeführten Jahren wurden in Deutschland jährlich im Mittel rund 781.000 Kinder geboren. Jedes Jahr kam für im Mittel etwa 155 dieser Kinder im erweiterten Neugeborenenscreening der Verdacht auf einen MCAD-Mangel auf. Im danach durchgeführten Konfirmationsscreening wurde dieser Anfangsverdacht für durchschnittlich 75 dieser 155 Kinder aufgrund von nach wie vor auffälligen Werten bestätigt, für 80 Kinder jedoch widerlegt, was den oben genannten 52% entspricht. In manchen Jahren war die Quote der bestätigten Fälle etwas niedriger, in anderen Jahren ein wenig höher, doch es bleibt festzuhalten, dass sich generell für etwa die Hälfte der hinsichtlich des MCAD-Mangels auffälligen Erstscreenings dieser Anfangsverdacht bereits mit dem Kontrollscreening wieder zerschlägt.
Um einen Irrtum, also eine falsch durchgeführte Analyse oder gar eine aufgrund von Schlampigkeit verwechselte Probe, wird es sich mit großer Sicherheit nicht handeln, aber es gibt doch einige Erklärungen für kurz nach der Geburt (leicht) erhöhte Werte. So weisen z.B. Frühgeborene oft erhöhte Acylcarnitinwerte auf, die den Verdacht auf einen MCAD-Mangel begründen könnten. Dies hängt auch mit der speziellen Frühgeborenennahrung zusammen, die in manchen Produkten, aufgrund der besseren Verdaubarkeit, einen hohen Anteil mittelkettiger Fette (MCT-Öl) enthält.
Sehr viel häufiger aber liegt die Situation vor, dass ein Kind tatsächlich von einem seiner Elternteile ein defektes MCAD-Gen vererbt bekommen hat. Damit ist es, genau wie der betreffende Elternteil, Anlagenträger oder “Carrier”. Es hat aber selbst keinen MCAD-Mangel, denn dieser setzt voraus, dass es von beiden Elternteilen − die dann beide zwingend zumindest Carrier sind − jeweils die defekte Genkopie vererbt bekommen hat. Eine einzige defekte Genkopie ist klinisch ohne Bedeutung, denn die vom anderen Elternteil geerbte intakte Kopie kann voll funktionsfähige MCAD-Enzyme in ausreichender Anzahl bilden. In den ersten Lebenstagen ist der Stoffwechsel eines Neugeborenen aber noch nicht vollständig eingespielt und wenn das Neugeborene dann noch relativ geringe Nahrungsmengen zu sich nimmt, kann es passieren, dass die auf einen MCAD-Mangel hindeutenden Blutbestandteile (“Acylcarnitine”) vorübergehend oberhalb des als unauffällig geltenden Normbereichs liegen. Dies kann vor allem dann auftreten, wenn das Neugeborene von Anfang an nur gestillt und nicht zugefüttert wird und das Nahrungsangebot somit während der ersten zwei bis drei Tage noch entsprechend knapp ist. Gerade bei Erstgeborenen dauert es üblicherweise ein paar Tage bis zum Milcheinschuss und aufgrund der bis dahin oft deutlichen Gewichtsabnahme des Neugeborenen, können die im Screening hinsichtlich des MCAD-Mangels getesteten Werte vorübergehend in leicht auffälliger Weise erhöht sein.
Wenn sich die aufgenommene Nahrungsmenge nach ein paar Tagen deutlich gesteigert hat und das Baby anfängt, wieder an Gewicht zuzulegen, normalisieren sich die bis dahin erhöhten Werte wieder, sodass meistens bereits das Kontrollscreening Entwarnung bringen kann.
In 48% der anfangs auffälligen Screenings zeigt allerdings auch noch die zweite Untersuchung weiterhin erhöhte Werte, was den Verdacht auf einen MCAD-Mangel erhärtet. Doch auch hier sind die Würfel noch nicht endgültig gefallen, denn es gibt unterschiedliche Defekte des MCAD-Gens, die zu unterschiedlich stark erhöhten Screeningwerten führen. Im ersten Moment – also bei der Bewertung des Screeningergebnisses – sind sie natürlich als “auffällig” zu betrachten. Bei weiteren Untersuchungen lässt sich die Schwere eines eventuellen MCAD-Mangels dann aber genauer klassifizieren. Bei vielen Defektvarianten hat z.B. entweder die von der Mutter oder vom Vater vererbte Anlage trotz vorliegendem Gendefekt immer noch einen gewissen Restnutzen, während einige andere Defekte zu einem völligen Funktionsverlust der betroffenen Genkopie führen. Auch letztere sind bei heterozygotem Vorliegen (nur von einem Elternteil vererbt) klinisch irrelevant, jedoch dauert die Einpendlung des Stoffwechsels, bis hin zur dauerhaften Normalisierung der Acylcarnitine, unter Umständen ein paar Tage länger, sodass sich die Werte vielleicht erst nach zwei bis drei Wochen im normalen Bereich bewegen. Bis der Stoffwechsel eines Neugeborenen wirklich in stabilen Bahnen verläuft, können generell bis zu vier Wochen vergehen. In jedem Fall ist ein hinsichtlich der mittelkettigen Acylcarnitine und deren Ratios (Quotienten) unauffälliger Befund − egal ob nach einer oder erst nach 10 Wochen − als Widerlegung des Anfangsverdachtes zu sehen, denn er drückt aus, dass der Stoffwechsel des Kindes auch in Hinsicht auf das MCAD-Enzym inzwischen rund läuft. Im Gegensatz dazu lassen sich bei Menschen mit tatsächlich als solchem zu bezeichnenden MCAD-Mangel zeitlebens die in charakteristischer Weise erhöhten Werte nachweisen. Selbst wenn bei ihnen in Zeiten optimaler Ernährung die reinen Acylcarnitinwerte (C8, C10, usw.) tatsächlich mal knapp innerhalb ihrer jeweiligen Normbereiche liegen können, zeigen doch die daraus gebildeten Ratios immer noch den Funktionseinbruch der Fettsäurenoxidation ab einer bestimmten Restkettenlänge an.
Kleiner Einschub: In den vergangenen Jahren haben mich – vermutlich aufgrund der vorangegangenen Abschnitte – schon oft Nachfragen von Eltern erreicht, in denen sie ihre Sorge schilderten, das Kontrollscreening (mit im Ergebnis zwar schon stark gesunkenen, aber weiterhin auffälligen Werten) sei vielleicht zu früh erfolgt, denn bestimmt wären die Werte noch ein paar Tage später, als es mit der Nahrungsaufnahme noch besser geklappt hat, dann völlig normal gewesen. Dazu muss ich leider sagen, dass die oben erwähnten ca. 52% der anfangs leicht erhöhten und bis zum Kontrollscreening bereits normalisierten Werte eben von Anfang an genau das waren: leicht erhöht! Nun hat man als gerade erst neu mit dem MCAD-Mangel konfrontierte Eltern selbstverständlich keine Vorstellung davon, was unter leicht, mittel oder stark erhöht zu verstehen ist. Auf diese Unterscheidung gehe ich weiter unter noch genauer ein, aber an dieser Stelle möchte ich nur kurz erwähnen, dass die auf einen Carrier-Status (also auf keinen MCAD-Mangel) hindeutenden Werte bei C8 (Octanoylcarnitin) meist deutlich unter 1 liegen, also gerade knapp über dem zugrunde gelegten oberen Grenzwert bis ungefähr 0.5 bis 0.7. Nur in diesen Fällen kommt es vor, dass bereits das Kontrollscreening (der “Recall”) nach ein paar Tagen unauffällige Werte zeigt, oder dass sich die Normalisierung der Acylcarnitinwerte in seltenen Fällen erst innerhalb der nächsten 2-4 Wochen einstellt.
Ist der C8-Wert im NG-Screening aber schon bei über 1, ist mit großer Wahrscheinlichkeit wenigstens ein milder, bei Werten von über 5 sogar ziemlich sicher ein klassischer MCAD-Mangel anzunehmen. In diesen Fällen ist auch mit noch so langem Abwarten keine vollständige Normalisierung der Acylcarnitinwerte zu erwarten. Eine weitere wichtige Rolle spielt der Quotient der Werte von C8 zu C10, der ausdrückt, wie stark der Funktionseinbruch bei der Aufspaltung der Fettsäurenketten mit 8er-Restlänge ausfällt. Bei reinen Carriern liegt dieser Quotient ungefähr bei 1 oder sogar noch darunter. Falls dir die Befundunterlagen des NG-Screenings vorliegen, kannst du es damit schon selbst überprüfen, oder schreib mich einfach mal an, dann können wir es uns zusammen anschauen.
Falls es nicht schon geschehen ist, steht als Nächstes eine weitere Blutentnahme für ein zweites Screening auf dem Plan. Das kann beim Kinderarzt, oder auch schon in der Stoffwechselambulanz der nächstgelegenen Uni-Klinik erfolgen. Möglicherweise werden zu diesem Zeitpunkt auch schon ein paar Milliliter Blut entnommen, die zur Durchführung einer molekulargenetischen Untersuchung benötigt werden. Meistens wird das aber erst dann gemacht, wenn auch das zweite Screening noch erhöhte Werte aufweist. Bei der genetischen Untersuchung − über deren Zweck die Eltern vom Arzt genau aufzuklären sind, und zu der sie ihr schriftliches Einverständnis geben müssen − wird innerhalb des DNA-Codes des MCAD-Gens nach bekannten, aber auch neuen Mutationen gesucht, die für die Verursachung des MCAD-Mangels infrage kommen. Es ist genau bekannt, an welchen Stellen der DNA sich die weit verbreiteten Mutationen befinden. Daher wird dort zuerst gesucht. Wird keine der bekannten Mutationen gefunden, erfolgt die Sequenzierung des gesamten Gens. Den Erfahrungen zufolge kann es einige Wochen oder sogar Monate dauern, bis das Ergebnis dieser Untersuchung feststeht. Erst alle diese Befunde zusammen werden letztlich ein relativ klares Bild darüber ermöglichen, ob bei deinem Kind tatsächlich ein MCAD-Mangel vorliegt, und falls ja, in welcher Ausprägung.
Seit einigen Jahren gehen auch mehr und mehr Stoffwechselzentren wieder dazu über, parallel zur molekulargenetischen Untersuchung auch die Restaktivität des MCAD-Enzyms anhand einer angelegten Zellkultur zu messen, um daraus Hinweise auf eine schwere, oder vielleicht doch nur leichte Ausprägung des MCAD-Mangels zu ermitteln. Diese Technik war schon früher mal erprobt, aber eine ganze Weile lang von vielen Uni-Kliniken vehement abgelehnt worden. Das lag daran, dass die Ergebnisse der früheren Analysemethoden sehr stark schwankten und sich auch in großen Bereichen überschnitten, sodass man den Resultaten kein großes Vertrauen schenken konnte. Inzwischen wurden aber mit großem Aufwand deutlich bessere und zuverlässigere Methoden zur Messung der “Residualaktivität” entwickelt, die sehr zuverlässig erkennen lassen, ob ein Kind einen schweren, einen milden, oder vielleicht sogar gar keinen MCAD-Mangel aufweist, wenn es nur Carrier ist. Sollte deine Stoffwechselambulanz diese Untersuchung nicht durchführen (wollen), kannst du dich diesbezüglich auch an eine andere Uni-Klinik wenden.
Zunächst einmal: Ruhe bewahren! Wie oben beschrieben, handelt es sich bisher lediglich um einen allerersten Verdacht. Der Tandem-Massenspektrograph, eine völlig ohne menschliches Zutun arbeitende computergesteuerte Anlage, hat die mit Fersenblut betropfte Trockenblutkarte deines Kindes analysiert und hinten eine Reihe von Zahlen ausgespuckt, verbunden mit der Feststellung, dass ein paar Werte außerhalb des Normbereichs liegen. Was bei deinem Kind tatsächlich vorliegt, weiß zu diesem Zeitpunkt noch niemand, denn bis zu einer auf sicheren Beinen stehenden Diagnose werden noch eine Reihe weiterer Untersuchungen notwendig.
Dennoch wirst du natürlich von Anfang an kein Risiko für dein Kind eingehen wollen! Das einzige, was du jetzt machen kannst, ist schlicht und einfach auf regelmäßiges Füttern zu achten. Die Empfehlungen bzgl. der maximalen Zeitabstände unterscheiden sich von Stoffwechselklinik zu Stoffwechselklinik teilweise deutlich. Ein ganz gutes Mittelmaß für die ersten Wochen ist sicherlich, die Mahlzeitenabstände 4 Stunden nicht überschreiten zu lassen. Früher geht immer! Vor allem dann, wenn du nicht nach starren Zeiten, sondern nach Bedarf stillen oder füttern willst, werden diese 4 Stunden Abstand ohnehin meist nicht erreicht. Mach dir aber aber bitte bzgl. dieser 4 Stunden keine zu großen Gedanken oder gar Sorgen. In diese und die für ältere Kinder empfohlenen maximalen Zeitspannen zwischen den Mahlzeiten, wurde von den Stoffwechselexperten ein großer Sicherheitspuffer einkalkuliert, sodass man sich bei strikter Beachtung dieser Zeiten immer weit im sicheren Bereich befindet. Im bildlichen Sinne balanciert man mit diesen 4 Stunden also beileibe nicht am Rande der Klippe, sodass man schon mit einem kleinen Fehltritt hinabstürzen könnte, sondern hält zu dem Klippenrand immer noch einen mehrere Meter breiten Sicherheitsabstand ein. Lies dazu am Besten auch den Artikel “Die Behandlung des MCAD-Mangels“
Ansonsten genieße dein Kind und beobachte es einfach mit etwas erhöhter Aufmerksamkeit. Bleierne Müdigkeit ist bei Neugeborenen ganz normal, aber wenn dir irgendetwas nicht mehr normal oder sogar besorgniserregend erscheint, dann zögere nicht, in der Stoffwechselambulanz der nächstgelegenen Uni-Klinik anzurufen und mit dem Hinweis, bei deinem Kind läge der Verdacht auf MCAD-Mangel vor, um Rat zu fragen.![]()
Die Diagnose des MCAD-Mangels ist wie ein aus vielen Teilen bestehendes Puzzlespiel. Erst wenn genügend Teile zusammengesetzt wurden, zeichnet sich ein einigermaßen klares Bild ab, ob tatsächlich ein MCAD-Mangel vorliegt, und welche Ausprägung dieser hat, also um welche Variante (mild oder schwer) es sich handelt.
Manchmal spricht schon das Ergebnis des Neugeborenenscreenings eine klare Sprache, in vielen Fällen mit grenzwertig auffälligen Analysewerten kann man aber erst nach weiteren Untersuchungen eine auf stabileren Beinen stehende Diagnose erstellen. An dieser Stelle möchte ich Dir schon mal eindringlich ans Herz legen, Dir grundsätzlich alle Befunde über die Untersuchungen deines Kindes, angefangen mit den Ergebnissen des Neugeborenenscreenings, von deinen Ärzten in Kopie aushändigen zu lassen. Lass dich niemals damit abspeisen, dass Du damit ohnehin nichts anfangen könntest und diese Befunde deshalb nicht bräuchtest! Wer keine Möglichkeit hat, die Ergebnisse selbst zu überprüfen, muss seinen Ärzten alles glauben − und leider kennen sich auch in den Stoffwechselzentren nur sehr wenige Ärzte wirklich soweit mit dem MCAD-Mangel aus, dass sie auch die Zahlen in den Befunden hinsichtlich des zu erwartenden Schweregrades interpretieren können. Während jedoch vor Gericht “im Zweifelsfall ist der Angeklagte freizusprechen!” gilt, wird es bei einem aufgekommenen Verdacht für das Vorliegen einer Stoffwechselstörung wie dem MCAD-Mangel genau andersherum gehandhabt: Entwarnung wird nur gegeben, wenn aus Sicht der behandelnden Ärzte zweifelsfrei nachgewiesen wurde, dass kein MCAD-Mangel vorliegen kann. Leider scheint für manche Ärzte der Fall bereits nach dem ersten auffälligen Befund des Neugeborenenscreenings vollkommen klar und das Interesse an weiterer Abklärung erloschen zu sein: wenn auffällige, wenn auch nur leicht erhöhte Werte festgestellt wurden, kann ja wohl irgendetwas nicht stimmen, also geht man doch besser gleich von einer schweren, also potenziell gefährlichen Variante des MCAD-Mangels aus. Sicher ist sicher − und dann erübrigen sich eigentlich auch weitere Untersuchungen zur genaueren Abklärung. Wenn die Eltern des Kindes zeitlebens vom schlimmsten Fall ausgehen, werden sie wenigstens nicht zu lasch − und fangen auch nicht an, irgendwann die Empfehlungen ihres Stoffwechselarztes womöglich in Frage zu stellen.
Wenn du an einen so denkenden Arzt gerätst, dann wechsele nach Möglichkeit die Stoffwechselambulanz, selbst wenn das einen etwas weiteren Anreiseweg bedeuten sollte. Der MCAD-Mangel ist eine im Leben des betroffenen Menschen zeitlebens präsente Stoffwechselstörung, die regelmäßige Kontrollen in der Uni-Klinik und daher ein auf lange Zeit angelegtes Arzt-Patienten-Verhältnis bedingt. Man sollte unbedingtes Vertrauen in seinen betreuenden Arzt setzen können, was aber voraussetzt, dass auch von ärztlicher Seite aus alles getan wird, um dieses Vertrauen aufzubauen. Eine “Ich weiß zwar auch nicht, was genau bei ihrem Kind vorliegt, aber wir tun einfach mal so, als ob!” -Haltung trägt nicht zur Vertrauensbildung bei, sondern erzeugt bei den Eltern das unangenehme Gefühl, von dem Arzt nicht ernst genommen zu werden. Ich weiß, wovon ich rede, denn spätestens nachdem unser erster Stoffwechselarzt (nach einigen Monaten mir zweifelhaft erscheinender Aussagen über die Diagnose) mich mit dem beinahe philosophisch anmutenden Satz “Das Einzige, was man mit Bestimmtheit über den MCAD-Mangel wissen kann, ist, dass man überhaupt nichts mit Bestimmtheit über den MCAD-Mangel wissen kann!” abzuwürgen versuchte, war mir endgültig klar, dass mich seine mehrfach erwähnte zwanzigjährige Erfahrung mit dem MCAD-Mangel zwar beeindrucken sollte, er in Wirklichkeit aber keine nennenswerte Ahnung davon hatte.
Wenigstens eine hieb- und stichfeste Diagnose des MCAD-Mangels sollte vorliegen, und zwar so, dass Ihr als Eltern diese Diagnose auch nachprüfen und nachvollziehen könnt. Dazu ist es − ich kann es wirklich nicht oft genug betonen − unbedingt notwendig, dass Ihr Euch von Beginn an (angefangen mit dem genauen Befund des NG-Screenings) alle Befundunterlagen in Kopie für eine eigene Akte aushändigen lasst.
Dann ist es “nur” noch notwendig, die in den Befunden genannten Zahlen und Begriffe auch zu verstehen. Dazu soll dieser Abschnitt Hilfestellung leisten.
Neugeborenenscreening:
Die Trockenblutkarte deines Kindes wurde als auffällig klassifiziert, weil einige Blutwerte erhöht waren, die auf das Vorliegen eines MCAD-Mangels hinweisen können. Leider gibt es keine scharfen Abgrenzungen zwischen den verschiedenen Bereichen, sondern mehr oder weniger große Überschneidungen, in denen eine Aussage bzgl. des zu erwartenden Gesamtergebnisses zu diesem frühen Zeitpunkt auf wackligen Beinen stehen würde. Allerdings gibt es − abgesehen von diesen Schnittmengen − doch Bereiche, in denen die gemessenen Werte schon sehr deutlich in eine bestimmte Richtung weisen.
Ein stark erhöhter C8-Wert (größer 5) und ein gleichzeitig stark erhöhter Quotient C8/C10 (größer 5) deutet mit sehr großer Wahrscheinlichkeit auf einen sich in den weiteren Untersuchungen herausstellenden klassischen, also schweren MCAD-Mangel hin. Vermutlich liegt die Mutation K329E (c.985a>g) in homozygoter Form vor, d.h. dein Kind hat diesen am häufigsten vorkommenden MCAD-Gendefekt von beiden Elternteilen vererbt bekommen. Selbst wenn sich in der molekulargenetischen Untersuchung zeigen sollte, dass wenigstens eine der beiden beteiligten Mutationen anders lautet, ist von einer vergleichbaren Auswirkung und somit auch Schwere auszugehen.
C8-Werte zwischen 0,7 und etwa 3 in Verbindung mit C8/C10-Ratios von 1,5 bis 3 legen die Aussicht auf eine milde(re) Ausprägung des MCAD-Mangels nahe. Untermauert würde diese Vermutung dann durch das Auffinden einer Mutationskombination, die als milde Form bekannt ist. Dies bedeutet, dass sie in den molekulargenetischen Untersuchungen relativ häufig gefunden wird, aber dass sie trotzdem noch nie zuvor im Zusammenhang mit erfolgten Stoffwechselentgleisungen zutage getreten ist. Für dein Kind werden seitens der Stoffwechselärzte dann zwar annähernd die gleichen Empfehlungen bzgl. maximaler Nüchterntoleranzzeiten und deines Verhaltens im Krankheitsfall gegeben, aber es kann schon beruhigend sein, zu wissen, dass noch nie zuvor ein Kind mit dieser Form des MCAD-Mangels jemals eine gefährliche Entgleisung erlitten hat − und das, obwohl die meisten Menschen mit dieser Ausprägung überhaupt nicht wissen, dass sie nach heutigen Erkenntnissen eigentlich einen MCAD-Mangel haben. Milde (med. “benigne” = gutartige) Ausprägungen des MCAD-Mangels werden hauptsächlich von compound-heterozygoten Mutationskombinationen hervorgerufen. Beide Eltern sind dabei Anlagenträger für unterschiedliche MCAD-Gendefekte und haben jeweils diesen Defekt an das Kind vererbt. Jede Mutation für sich kommt deshalb nur einmal vor (heterozygot), allerdings gemeinsam (compound) mit der anderen.
Nur gering über dem Normbereich liegende C8-Werte (kleiner 0,7) und gleichzeitig kleine C8/C10-Ratios (kleiner 1,5) sind ein starkes Indiz für einen falsch-positiven Befund. Mit hoher Wahrscheinlichkeit liegt bei deinem Kind ein heterozygoter MCAD-Gendefekt vor, d.h. es hat ihn entweder von der Mutter oder vom Vater geerbt, aber nicht von beiden. Somit ist dein Kind − so wie der betreffende Elternteil − nur Anlagenträger (“Carrier”), aber nicht selbst von einem MCAD-Mangel betroffen. Möglicherweise haben sich die in diesem Befund noch auffälligen Werte beim Kontrollscreening, oder bei einer wenige Wochen später durchgeführten Blutuntersuchung schon vollkommen normalisiert. Dies wäre dann als eindeutige Widerlegung des Anfangsverdachts zu werten. Die vermutlich ebenfalls noch durchgeführte Genanalyse kann dann mit dem Auffinden einer einzigen Mutation noch den letzten Beweis für den reinen Carrier-Status deines Kindes erbringen.
Alle weiteren Werte liegen in den Überlappungsbereichen zwischen Carrier und milder Form, bzw. zwischen milder und schwerer Form, sodass sich erst mit den Ergebnissen der weiteren Befunde ein deutlicheres Bild ergeben wird.
Einflussgrößen
Wie auch beim folgenden Kontrollscreening, kann es schon im Neugeborenenscreening Störfaktoren bzw. besondere Umstände geben, die Auswirkungen auf die bei dem Neugeborenen gemessenen Werte haben, sodass z.B. trotz eines vorliegenden schweren MCAD-Mangels nur geringfügig erhöhte Acylcarnitinwerte gefunden werden, die zunächst mal auf einen milden MCAD-Mangel hindeuten können. Dies kann beispielsweise passieren, wenn ein Kind aufgrund eines deutlich zu frühen Geburtstermins, einer Neugeboreneninfektion oder irgendeines anderen Umstands, von Beginn an mit Infusionen versorgt wird. Dadurch kann sich in der Zeit bis zur Blutentnahme gar keine so deutliche katabole Stoffwechsellage einstellen, wie es bei einem von Anfang an ausschließlich gestillten Kind der Fall ist. Das Neugeborene verbraucht aufgrund der Infusionen kaum Körperfett und die Acylcarnitinwerte sind deshalb nur geringfügig erhöht. Der MCAD-Mangel wird aufgrund der trotzdem etwas erhöhten C-Werte gefunden, aber die Werte aus dem NG-Screening-Befund lassen noch keine zuverlässige Voreinschätzung in schwere oder milde Variante zu. Umgekehrt kann ein Frühchen, dem eine spezielle, leichter verdauliche Frühgeborenennahrung zugeführt wird (diese enthält oft einen höheren Anteil an mittelkettigen Fetten) erhöhte C8-Werte aufweisen, die fälschlicherweise den Verdacht auf einen MCAD-Mangel nahelegen, obwohl das Kind vielleicht dann doch nur Carrier ist.
Auch können Neugeborene von sich überwiegend vegetarisch oder vegan ernährenden Müttern aufgrund dieses Umstands selbst ebenfalls einen erniedrigten Carnitinspiegel haben, usw…
Die zuverlässigsten Vorabeinschätzungen lassen sich aus den Screening-Befunden ableiten, wenn das Kind in der Zeit bis zur Blutentnahme gesund war, ausschließlich gestillt wurde, und dabei erwartungsgemäß erst einmal ein paar hundert Gramm Gewicht verloren hat. Die größte Gewichtsabnahme, und somit auch die schon deutlichsten Screeningwerte sind bei Erstgeborenen zu erwarten, da bis zum richtigen Milcheinschuss bei der Mutter dann noch 2-3 Tage vergehen, und das Neugeborene somit besonders stark von seinen Reserven zehrt. Fast immer lässt sich dann schon aus dem ersten Screening eine ganz deutliche Eingruppierung in Carrier, milden oder schweren MCAD-Mangel ableiten, die von den folgenden Untersuchungen nur noch weiter bestätigt wird.
Kontrollscreening und weitere Acylcarnitinprofile:
Im zweiten Screening und allen weiteren Blutuntersuchungen wird der im NG-Screening noch stark erhöhte C8-Wert schon deutlich abgesunken sein. Selbst C8-Werte im Bereich von größer 10 können nach einigen Tagen schon nur noch 2-3 betragen. Dies ist für den MCAD-Mangel völlig normal, sobald das Kind anfängt größere Mengen an Nahrung zu sich zu nehmen. Im Gegensatz dazu bleibt aber der Quotient C8/C10 über einen langen Zeitraum annähernd konstant. Dies sagt aus, dass nun zwar insgesamt weniger Fettsäuren in den Zellen verwertet werden, als in den ersten Tagen aufgrund der zu dem Zeitpunkt noch bestehenden Nahrungsknappheit (daraus ergibt sich der niedrigere C8-Wert), aber dass es auch auf diese geringere Menge an Fettsäuren bezogen, bei deren Verwertung einen starken Funktionseinbruch bei einer Kettenrestlänge von 8 Kohlenstoffmolekülen gibt. Ein somit nach wie vor deutlicher Indikator für einen schweren, oder auch milden MCAD-Mangel.
Es gelten die gleichen Unterteilungen, wie im Abschnitt “Neugeborenenscreening”, allerdings mit Anpassung der C8- und Beibehaltung der C8/C10-Werte.
Manche MCAD-Ausprägungen lassen sich trotz aller möglichen Untersuchungen nicht klar einordnen, sondern bleiben in einem dieser Graubereiche. In diesen Fällen sollte man zur Sicherheit des Kindes tatsächlich von der jeweils schwereren Alternative ausgehen und entsprechend vorsichtig und aufmerksam vorgehen.
Wenn dagegen tatsächlich alle Befunde eindeutig auf das Vorliegen eines reinen Carrier-Status und somit keinen MCAD-Mangel hinweisen, gibt es auch keinen Grund, ein solches Kind zwanghaft krank zu reden. Sollte sich der betreuende Stoffwechselarzt nicht auf diese Sichtweise einlassen wollen (teilweise zum reinen Selbstschutz), habt ihr als betroffene Eltern immer die Möglichkeit, euch zwecks zweiter Meinung an eine andere Stoffwechselambulanz zu wenden. In Deutschland ist man zum Glück (noch) nicht auf Gedeih und Verderb an den ersten Arzt, den man aufgesucht hat, gebunden, sondern hat die freie Wahl.
![]()
Die Diagnose eines milden MCAD-Mangels basiert auf zwei wesentlichen Feststellungen als Resultat der erfolgten Untersuchungen:
Bei seltenen oder bislang sogar noch ganz unbekannten neuen Mutationen im für das MCAD-Enzym zuständigen ACADM-Gen des Kindes, kann man anhand verschiedener Computerprogramme simulieren und berechnen lassen, was diese Mutationen mit dem MCAD-Enzym anstellen und wie stark die davon verursachten Einschränkungen sein könnten. Die in solchen Fällen aber deutlich bessere Diagnosemethode ist tatsächlich, die Leistungsfähigkeit dieses Enzyms mittels einer Residualaktivitätsanalyse in den Lymphozyten (weißen Blutkörperchen) überprüfen zu lassen.
Besonders die Feststellung, dass es bei Kindern mit der gleichen Mutationskombination noch nie zu einer Stoffwechselentgleisung gekommen ist − und das selbst zu Zeiten, als noch nicht frühzeitig gescreent wurde, und somit die Eltern in Krankheitsphasen auch nicht peinlich genau auf regelmäßiges Füttern ihrer Kinder achteten, und kein Mensch daran dachte, bei Nahrungsverweigerung zwecks Infusion in die Klinik zu fahren − sollte geeignet sein, dir ein paar der momentan vielleicht noch bestehenden Sorgen über die Zukunft deines Kindes zu erleichtern.
Mit der schriftlichen Mitteilung, dass die Befunde deines Kindes auf eine milde Variante des MCAD-Mangels hindeuten, und der sich daraus möglicherweise für dich selbst ergebenden psychischen Beruhigung, ist es dann aber leider auch schon getan! In Bezug auf die Behandlungen durch die euch betreuenden Ärzte in der Stoffwechselambulanz, die regelmäßigen Kontrolltermine und ihre Empfehlungen hinsichtlich der Nüchterntoleranzzeiten (also der maximalen Abstände zwischen den Mahlzeiten des Kindes), der eventuellen täglichen Zuführung von Carnitin und dem Rat, bei Anzeichen von Nahrungsverweigerung sofort in die Klinik zu kommen, um das Kind während dieser Phase per Glukoseinfusion ernähren und somit bloß nicht hungern zu lassen, ändert sich nämlich mit großer Wahrscheinlichkeit überhaupt nichts. Eine hundertprozentige Sicherheit für die Unbedenklichkeit einer bestimmten MCAD-Variante gibt es nämlich nicht, und wird es mit noch so vielen unauffälligen Beispielen auch niemals geben. Während es nämlich sehr einfach festzustellen ist, ob eine bestimmte Mutationskombination ein Risiko birgt (ein einziger bestätigter Entgleisungsfall reicht aus, um diese Mutationskombi als riskant einzustufen), funktioniert dies in entgegengesetzter Richtung leider nicht.
Als Beispiel sei die häufige MCAD-Genmutation c.199t>c (p.Y67H) genannt. Diese Mutation − und viele andere − wurde erst mit der Einführung des erweiterten Screenings gefunden, und war bis dahin völlig unbekannt. Dies bedeutet aber nicht, dass es sich dabei erst um einen in den letzten Jahren verbreiteten MCAD-Gendefekt handelt. Aufgrund der Häufigkeit, mit der diese Mutation inzwischen in homozygoter Form (c.199t>c/c.199t>c) und in compound heterozygoter Form (vor allem in Kombination mit der als riskant bekannten Mutation K329E / c.985a>g) gefunden wird, kann man davon ausgehen, dass sie schon seit mehreren hundert Jahren, also über viele Generationen hinweg, im Umlauf ist und heute sehr viele Menschen Carrier für diese Genmutation sind – wenn auch natürlich längst nicht so viele, wie für die häufigste Mutation c.985a>g.
Schon vor 2004 durchgeführte genauere Analysen dieser Mutation und ihrer Auswirkungen haben gezeigt, dass die Funktion des entstehenden MCAD-Enzyms anscheinend nur geringfügig beeinflusst wird, und das Enzym unter normalen Bedingungen fast ohne Einschränkung arbeitet. Wie es unter Krankheitsbedingungen aussieht, wird in letzter Zeit vermehrt wissenschaftlich untersucht, mit dem Ziel in Zukunft vielleicht sogar für Kinder, bei denen diese Mutation selbst in Kombination mit K329E gefunden wird, die Diagnose MCAD-Mangel wieder vollständig fallen lassen zu können. Bis zu gesicherten Studienergebnissen wird allerdings noch einige Zeit vergehen. In der in den letzten Jahren immer häufiger durchgeführten Residualaktivitätsmessung wurde bei Kinder mit der Mutationskombination c.985a>g/c.199t>c eine Aktivität von um die 20% gemessen. Da aus dem Gen mit der schweren Mutation c.985a>g nur Enzyme mit keiner nennenswerten Aktivität (weniger als 0,5%) produziert werden können, wird fast die gesamte Leistung von den aus dem Gen mit der Mutation c.199t>c gebauten MCAD-Enzymen erbracht. Bei homozygotem Vorliegen von c.199t>c würde somit eine Aktivität von sogar etwa 40% feststellbar sein, was dann bereits im Bereich der bei Carriern messbaren Aktivitäten läge. Allerdings ist diese homozygote Kombination dann doch wieder so selten, dass sie in den letzten Jahren anscheinend noch nicht im Rahmen einer Residualaktivitätsmessung erfasst wurde. Somit kann man bislang nur den Schluss ziehen, dass aufgrund des bisherigen Fehlens erfasster Fälle echter Stoffwechselentgleisungen bei Menschen mit der zuvor genannten compound heterozygoten Variante dieser Mutation, selbst die bei ihnen vorliegenden “nur” 20% Restaktivität der MCAD-Enzyme bereits ausreichen dürften, um einen weitgehend normalen Ablauf des Stoffwechsels zu ermöglichen.
Momentan lässt sich ein hieb- und stichfester Beweis dafür leider noch nicht erbringen, denn dazu müsste man Kinder mit diesem Gendefekt in homozygoter oder compound-heterozygoter Form gezielt über einen längeren und normalerweise für den MCAD-Mangel kritischen Zeitraum hinweg hungern lassen. Dieses Risiko werden aber weder Ärzte noch Eltern eingehen wollen, und selbst wenn in einer Studie mit 100 Kindern herauskäme, dass es bei einem solchen Fastentest zu keinen MCAD-typischen Auffälligkeiten oder gar einer Entgleisung kam, würde dies nicht als Garantie dafür ausreichen, dass auch dem 101. oder 102. Kind in ähnlicher Situation nichts passieren könnte.
Im Allgemeinen, d.h. mit Ausnahme dieser einen Mutation Y67H (c.199t>c), basiert die (vorsichtige) Diagnose “milder” MCAD-Mangel daher auch nicht auf den ermittelten Mutationen, sondern auf dem klinischen Gesamtbild eines Kindes. Dazu zählt u.a. wie lang das Kind nüchtern sein kann, bevor es zu einer feststellbaren Unterzuckerung oder anderen klinischen Symptomen kommt, ob es die üblichen Kinderkrankheiten ohne spezielle Maßnahmen überstanden hat, oder ob es aufgrund einer Erkältung schon mal ins Krankenhaus musste. Braucht es eine kohlenhydratreiche Nachtmahlzeit, um morgendliche Unterzuckungen zu verhindern, oder übersteht es die Nächte auch ohne Nachtmahlzeit problemlos? Aber selbst dieses klinische Gesamtbild lässt sich nicht ohne weiteres aufstellen, denn die wichtigste Maßnahme beim MCAD-Mangel ist und bleibt nun mal die Vermeidung jedes noch so kleinen Risikos.
Ein als mild (med. “benigner”=gutartig) eingestufter MCAD-Mangel wird daher seitens der Stoffwechselärzte identisch wie eine klassische, d.h. für ihr Entgleisungsrisiko bekannte Variante behandelt. Je nach behandelndem Arzt, und dabei hängt es wirklich rein von seiner persönlichen Meinung ab, wird vielleicht noch die maximale Nüchterntoleranzzeit um 1 bis 2 Stunden erhöht, aber auch diese soll dann nicht überschritten werden. Es wird weiterhin regelmäßige Kontrolluntersuchungen in der Stoffwechselambulanz geben, vielleicht mit etwas größeren zeitlichen Abständen, als bei Kindern mit den schweren MCAD-Varianten. Und auch im Fall der Nahrungsverweigerung, oder des wiederholten Erbrechens sollen sich die Eltern, aufgrund der nicht 100%igen Unbedenklichkeitsgarantie dieser milden Ausprägung, auf kein riskantes Spiel einlassen, sondern ihr Kind lieber in der Klinik per Infusion über die vielleicht doch nicht ganz unkritische Zeit bringen lassen.
Auf diese Weise wird sich natürlich leider auch nicht feststellen lassen, ob dieser ganze “Aufwand” tatsächlich gerechtfertigt ist, oder das Kind − und damit auch die ganze Familie − auch ohne die nächtlichen Fütterungen, Carnitingaben und zeitweisen Klinikaufenthalte problemlos und weitgehend unbelastet leben könnte. Von ärztlicher Seite aus geht es ausschließlich um Risikominimierung − nicht nur für die Kinder, auch für sich selbst, denn sie werden zur Rechenschaft gezogen werden, wenn sich ihre Empfehlungen im Nachhinein als Fehler herausstellen! Die Verantwortung für ihr Kind tragen aber letztlich die Eltern, und sie werden im Laufe der Zeit ein feines Gespür dafür entwickeln, wie sich ihr Kind in dieser oder jener Krankheitssituation verhält, wann sie die Lage selbst im Griff zu haben glauben, und wann sie sich doch wohler fühlen, wenn ihr Kind zur Sicherheit in der Klinik versorgt wird.
Auf gar keinen Fall! Ein diagnostizierter MCAD-Mangel ist ein MCAD-Mangel, egal ob er als mild oder als normal eingestuft wird! Es gibt zwar die begründete Vermutung, jedoch keine Garantie, dass es bei einer als mild angenommenen Variante niemals zu einer Entgleisung kommen wird. Eine Ausnahme sind laut einer Studie der Uni-Klinik Düsseldorf aus dem Jahr 2012 diejenigen Varianten, bei denen wenigstens eine Y67H- und der E43K-Mutation beteiligt ist. Deren Residualaktivitäten liegen in Kombination mit der schweren K329E üblicherweise bei ungefähr 20% (Y67H), oder sogar bei über 30% (E43K). Aber auch, wenn man wie in dieser Studie zu dem Ergebnis kommt, dass man den davon betroffenen Kindern genaugenommen keinen MCAD-Mangel mehr anhängen müsste, bedeutet dies noch lange nicht, dass sich diese “interne” Erkenntnis auch auf die in Deutschland praktizierte Behandlung der MCAD-Patienten auswirkt. Inzwischen wurden sogar noch eine ganze Reihe weiterer Mutationskombinationen identifiziert, deren Enzymaktivitäten im Bereich größer als 20% gemessen wurden, sodass man auch für diese eigentlich eine entsprechende Unbedenklichkeit wie für die beiden zuvor genannten Varianten annehmen könnte. Leider sind die meisten der neu entdeckten Mutationskombinationen aber so selten, oftmals nur Einzelfälle, dass sich viele Stoffwechselärzte in diesem Punkt lieber nicht festlegen wollen und deshalb die Behandlung weiterhin wie bei einem schweren MCAD-Mangel angehen.
Es gibt bei den unterschiedlichen Stoffwechselstörungen die verschiedensten Trigger, die eine Krise auslösen können − und kein Kind ist wie das andere. Trigger können z.B. Fiebertemperaturen, körperliche Erschöpfungszustände nach anstrengendem Sport, äußere Temperaturen, irgendwelche Medikamenteneinnahmen, Nahrungsmittel, u.v.m. sein. Z.B. könnte eine nach vielen Jahren erstmals auftretende extreme Sommerhitze bei einem bis dahin völlig unauffälligen Kind mit vermutetem mildem MCAD-Mangel als Auslöser für eine beginnende Entgleisung wirken, während ein anderes Kind mit der gleichen Mutationskombination diese Temperatur ohne die geringsten Probleme wegsteckt, aber vielleicht Jahre später auf einen ganz anderen Trigger erstmals auffällig reagiert.
Was für eine positive psychische Wirkung man als Eltern aus der Erkenntnis gewinnt, dass es sich um eine (vermutlich) milde Variante handelt, bleibt letztlich jedem selbst überlassen. Vielleicht wirkt alleine schon das Wissen etwas beruhigend, dass zumindest keine der bereits durch eindeutige Entgleisungen in Erscheinung getretenen Mutationskombinationen vorliegt. Im Interesse des Kindes darf eine solche Mitteilung jedoch niemals dazu führen, dass die nun mal diagnostizierte Stoffwechselstörung seitens der Eltern (und auch der behandelnden Ärzte!) nicht ernst genommen wird!
Vielleicht wird die medizinische MCAD-Forschung in einigen Jahren soweit sein, dass die Schwellwerte, ab denen ein Screeningergebnis oder ein Acylcarnitinprofil als auffällig gilt, angehoben werden, oder die Datenbasis für einzelne Mutationskombinationen deutlich angewachsen sein. Vielleicht werden dann einige der heute noch mit “milder, aber behandlungsbedürftiger MCAD-Mangel-Ausprägung” diagnostizierte Kinder, dann schon im NG-Screening als unauffällig gelten, und überhaupt keine MCAD-Verdachts-Mitteilung erhalten. Aber bis dahin ist ein diagnostizierter MCAD-Mangel, und sei es auch eine als noch so mild angenommene Variante, dennoch ein MCAD-Mangel, der seitens der Eltern jederzeit ernst genommen und seitens der Ärzte in den Kliniken immer gemäß der im Notfall-Ausweis beschriebenen Maßnahmen behandelt werden muss.![]()
Vor der Einführung des “Erweiterten Neugeborenenscreenings” wurde der MCAD-Mangel ausschließlich bei Kindern, Jugendlichen und Erwachsenen diagnostiziert, die aufgrund schwerster Krankheitssymptome, deren Ursache zunächst meist völlig unklar war, ins Krankenhaus eingeliefert worden waren. Auch bei einer Anzahl von Fällen von plötzlichem Kindstod (wenn die Todesursache unbekannt ist, wird immer eine Obduktion zur genauen Feststellung der Todesumstände durchgeführt), wurde im Nachhinein ein klassischer MCAD-Mangel festgestellt.
Bei der Suche nach Gendefekten, die als Verursacher dieser schweren metabolischen Stoffwechselentgleisungen infrage kämen, stießen die Wissenschaftler fast immer auf die gleiche Genmutation im für die Bildung des MCAD-Enzyms zuständigen ACADM-Gen (ACADM=”Acyl-CoenzymA Dehydrogenase, Mittelkettig”).
Diese in fast allen Fällen gefundene Mutation c.985a>g (andere Bezeichnung: K329E) führt nach den Erkenntnissen der letzten 40 Jahre vor allem bei homozygotem Vorliegen − wenn ein Kind diese Mutation sowohl von seiner Mutter, als auch von seinem Vater vererbt bekommen hat − zu einer schweren Form des MCAD-Mangels. Ein solches Kind ist extrem gefährdet, eine Stoffwechselentgleisung zu erleiden, wenn es − besonders in Krankheitszeiten mit Nahrungsverweigerung − zu große Mahlzeitenpausen macht, oder aufgrund von Magen-Darm-Infekten mit Erbrechen oder Durchfall zu wenig Energie in Form von Kohlenhydraten bei sich behalten und sinnvoll verwerten kann. Warum es aus dem Fettanteil der aufgenommenen Nahrung keinen großen Nutzen ziehen kann, lies bitte in den MCAD-Informationen in dem Artikel “Was ist ein MCAD-Mangel?” nach, denn hier würde dies zu weit führen.
Im ersten Moment wird dich das Lesen von so schlimmen und manchmal sogar tödlich endenden Entgleisungen sicherlich erschrecken, aber dein Kind ist schon jetzt viel besser dran! Ihr als Eltern wisst nämlich bereits, dass − obwohl man es eurem Kind nicht ansehen kann − da doch irgendetwas an ihm ist, was unter bestimmten Voraussetzungen ein hohes Gesundheitsrisiko für euer Kind darstellen kann.
Zu allen diesen schlimmen Entgleisungen mit teilweise tödlichem Ausgang in der Vergangenheit kam es nur deshalb, weil die Eltern dieser Kinder nicht das Geringste von dem bei ihnen vorliegenden MCAD-Mangel ahnten. Hätten Sie, oder die Ärzte der Notfallaufnahmen etwas geahnt, wäre es wahrscheinlich in allen diesen Fällen gut ausgegangen, bzw. in den meisten Situationen gar nicht erst zu einer Entgleisung gekommen. Dies ist jedenfalls eine der wesentlichen Erkenntnisse, zu der eine Langzeituntersuchung zum MCAD-Mangel schon gekommen ist.
Die Maßnahmen zur Vermeidung einer solchen Notfallsituation sind einfach und mit wenigen Worten auf den Punkt gebracht:
Das ist in einem kurzen Satz eigentlich schon alles, was es zu beachten gilt! Alles Weitere sind Feinheiten, wie dieses Ziel in der einen oder anderen Situation zu erreichen ist.
Die für dein Kind in seinem jeweiligen Alter geltenden maximalen Mahlzeitenabstände, die es aus Sicherheitsgründen nicht zu überschreiten gilt, werden dir vermutlich schon von der Stoffwechselambulanz mitgeteilt worden sein. Ansonsten kannst du im Artikel “Wie wird der MCAD-Mangel behandelt?” eine Tabelle mit Zeitvorschlägen finden, die unter den deutschen Stoffwechselambulanzen weit verbreitet sind.
Nachts empfiehlt es sich einen Wecker zu stellen, um dein Kind nach spätestens diesem empfohlenen Zeitraum wieder zu stillen, oder mit einem Fläschchen zu füttern.
Keine Angst! Die in der Tabelle aufgeführten Zeiten sind immer noch sehr kurz gewählt, um auf jeden Fall auf der sicheren Seite zu bleiben. Nach einer ordentlichen Mahlzeit kommt dein Kind bei normalem Gesundheitszustand besonders nachts auch locker noch ein paar Stunden länger ohne erneutes Füttern aus, ohne dass sich bei ihm gleich eine Entgleisung anbahnt. Fast alle in der Vergangenheit bei unbekanntem MCAD-Mangel erfolgten nächtlichen Entgleisungen traten nämlich im Zusammenhang mit einem Magen-Darm-Infekt auf, wenn das Kind schon am Abend keinen Appetit mehr hatte, oder seine letzte Mahlzeit fast vollständig wieder erbrochen hatte. Während der vorangegangenen Monate oder gar Jahre hatten diese Kinder auch keine Probleme mit den nächtlichen Mahlzeitenpausen. Oftmals sahen es die Eltern ihrem Kind beim Zubettbringen aber auch noch gar nicht an, dass es etwas ausbrütete! Daher ist es wichtig, bei der Beachtung dieser Zeiten nicht nachlässig zu werden.
Für euch als Eltern werden die Nächte mit einem zuverlässig klingelnden Wecker aber sehr viel entspannter verlaufen. Fast alle Eltern machen trotzdem bereits innerhalb der ersten Monate die Erfahrung, wie schrecklich man sich fühlt, wenn man irgendwann mal vor lauter Müdigkeit den klingelnden Wecker einfach abschaltet und sofort wieder einschläft − und dann aus dem Schlaf aufschreckt, um voller Panik festzustellen, dass man den spätestens fälligen Fütterungszeitpunkt bereits um eine oder mehr Stunden überschritten hat. Daher empfiehlt es sich wirklich, nicht nur einen, sondern sogar zwei Wecker kurz nacheinander klingeln zu lassen. Alternativ kann man auch den Wecker so weit weg platzieren, dass man sich zum Abschalten vollständig aus dem Bett erheben muss. ![]()
Sofern aus den Befunden zweifelsfrei hervorgeht, dass tatsächlich ein MCAD-Mangel vorliegt, spielt diese Frage nach der Schwere bzw. der Variante vordergründig nur eine ganz untergeordnete Rolle. Wie oben bei der Frage nach der Bedeutung einer milden Variante beantwortet wurde, macht die Differenzierung zwischen mild und schwer hinsichtlich der Behandlung des Kindes durch den Stoffwechselarzt, die euch genannten maximalen Fütterungsintervalle, sowie alle weiteren Vorsichtsmaßnahmen, keinen wesentlichen Unterschied. Sehr viel mehr als einen erleichterten Stoßseufzer und eine kleine Reduzierung der besonders am Anfang oft noch großen Ängste und Sorgen um die Zukunft des Kindes, hat man als Eltern nicht davon. Dennoch kann genau dies schon eine gewaltige psychische Entlastung für die so kurz nach der Geburt mit einer so erschreckenden Nachricht konfrontierten Eltern bedeuten. Daher ist es ein verständlicher und begründeter Wunsch der Eltern, möglichst ausführlich zu erfahren, was es mit dem MCAD-Mangel des eigenen Kind nun genau auf sich hat.
Leider können viele Ärzte diesen Wunsch nicht nachvollziehen, sondern haben die Haltung “Da sich an der Behandlung ohnehin nichts ändern wird, spielt es für mich − und damit auch für die Eltern − überhaupt keine Rolle, ob es sich um eine schwere oder möglicherweise milde Form handelt.” So ließ sich auch unser eigener Stoffwechselarzt − trotz eines über alle Befunde hinweg eindeutig (mit ein wenig Erfahrung!) erkennbaren Carrierstatus meines Sohnes − nicht von seiner Haltung abbringen, dass wir der Einfachheit halber mal von einem schweren MCAD-Mangel ausgehen sollten.
Aber bloß weil der Stoffwechselarzt den Eltern gegenüber sagt “Ob es sich eher um eine schwere oder eine milde Form handelt, braucht Sie nicht zu interessieren!”, bedeutet das nicht, dass sich dieser Wunsch nach detaillierterer Information und das Bedürfnis, endlich etwas mehr Klarheit über den genauen Befund des Kindes zu bekommen, einfach so im Gehirn der Eltern ausknipsen lässt!
Sollte es dir genauso gehen, bleibt dir eigentlich nur, dich selbst intensiver mit dem Thema zu befassen, um dir diese und andere Fragen irgendwann selbst beantworten zu können. Das erfordert aber einigen Aufwand und persönlichen Einsatz, denn alleine schon die Frage nach der anzunehmenden Schwere/Milde des bei deinem Kind vorliegenden MCAD-Mangels lässt sich nicht so ohne weiteres beantworten! Hierzu müssen alle vorliegenden Befunde in ihrer Gesamtheit betrachtet werden. Angefangen mit dem Bericht des Neugeborenenscreenings, über die Ergebnisse des/der Kontrollscreenings, bis hin zu den in der molekulargenetischen Untersuchung gefundenen Mutationen, und ggf. auch noch die ermittelten Restaktivitäten des Enzyms.
Je mehr Befunde vorliegen, desto deutlicher wird das sich daraus ergebende Gesamtbild. Allerdings erfordert es auch noch etwas Erfahrung, um diese Zahlen in ihrer Gesamtheit zu deuten, und selbst dann wird die Frage nach der groben Eingruppierung des vorliegenden MCAD-Mangels in manchen Fällen letzten Endes unbeantwortet bleiben müssen.
Manchmal erhält man von seinem Stoffwechselarzt leider keine genauere Erläuterung der Befunde. Manche Ärzte können mangels eigener Erfahrung selbst nichts mit den darin enthaltenen Zahlenwerten anfangen und wieder andere (zum Glück nicht viele) Ärzte sind anscheinend der Meinung, dass es Zeitverschwendung für sie wäre, den Eltern die Befunde zu erläutern, da diese ohnehin nicht in der Lage seien, diese Erläuterungen zu verstehen. Falls du keine zufriedenstellenden Informationen zu den Befunden deines Kindes erhalten hast, kannst du mir die dir vorliegenden Zahlen gerne per Email an schicken. Dann können wir mal darüber reden, bzw. schreiben.
Kleiner Einschub meiner persönlichen Erfahrung:
Ich selbst habe die ersten vier Wochen lang meinem Stoffwechselarzt jedes Wort geglaubt, weil seine eigene Aussage, er habe seit 20 Jahren Erfahrungen mit dem MCAD-Mangel und kenne sich deshalb bestens aus, natürlich keinen Widerspruch zuließ und überdies auch tatsächlich eine Menge Vertrauen einflößte. Ich missachtete allerdings (sehr zu seinem Unwillen) seine mit Nachdruck ausgesprochene Empfehlung, mich nicht selbst mit dem Thema zu beschäftigen. Im zweiten Monat kamen mir aufgrund der vorliegenden Befunde aus NG-Screening, Kontrollscreening und molekulargenetischer Analyse und der darin genannten Zahlen erste Zweifel an der Korrektheit seiner Aussage, mein Kind habe einen schweren MCAD-Mangel, denn für mein Empfinden drückten die Zahlen und die Formulierungen doch sehr deutlich aus, dass es Anzeichen für einen reinen Carrierstatus oder allerhöchstens einen sehr milden MCAD-Mangel gäbe. Auf die entsprechende Nachfrage hin folgte allerdings nur die Erwiderung, dass ich aufhören solle, mich damit zu beschäftigen, denn das nütze meinem Kind überhaupt nichts! Er kenne sich aus, und es sei ja wohl irgendetwas nicht in Ordnung, folglich gehe er von einem schweren MCAD-Mangel aus. Ich wollte mich damit im Interesse meines Kindes aber nicht abspeisen lassen, und forschte die folgenden fünf Monate lang täglich viele Stunden lang in allen im Internet zu findenden medizinischen Berichten zum MCAD-Mangel nach, was die genannten Zahlen genau zu bedeuten hatten. Nach über 700 Stunden, die in die Recherchen und das Zusammentragen der Informationen auf dieser Seite eingeflossen sind, war ich zu 99% überzeugt, dass mein Kind gemäß der Darstellung der Befunde keinen MCAD-Mangel haben könne. Es stand allerdings nirgendwo so konkret drin und der Arzt war auch nicht bereit, sich meine Argumentation anzuhören, geschweige denn von seiner eigenen Darstellung abzugehen. Als wir beschlossen, zwecks Einholung einer zweiten Meinung an eine andere Uni-Klinik zu wechseln, forderte ich von seinem Sekretariat noch einmal einen vollständigen Satz an Kopien der vorliegenden Befunde an − außer natürlich der handschriftlichen Notizen, in denen er mit Sicherheit vermerkt hatte, dass man mit mir nicht vernünftig reden könne. In einem ihm bereits seit dem zweiten Lebensmonat meines Kindes vorliegenden Zusatzbefund zur molekulargenetischen Untersuchung stand als Abschlussergebnis drin, dass alle erfolgten Untersuchungen zeigten, dass ganz eindeutig nur eine heterozygote Genmutation vorliege und daher ein MCAD-Mangel ausgeschlossen werden könne. Von diesem Befund hatten wir die gesamte Zeit über nicht das Geringste erfahren. Eigentlich wäre das ganze Thema bereits nach zwei Monaten für uns erledigt gewesen.
Eine ähnliche Erfahrung haben in den vergangenen Jahren noch einige andere Familien aus dem früheren MCAD-Forum gemacht. Es waren zwar in der Tat nur ganz wenige, nämlich insgesamt 17 von rund 550 Familien (davon 7 alleine nur in den Jahren 2020-2023!), bei denen sich der absolut sichere MCAD-Mangel nach meiner persönlichen Einschätzung der Befundwerte und der danach erfolgten Einholung einer Zweitmeinung in einer anderen Stoffwechselklinik als völlige Fehldiagnose herausstellte. Darüber hinaus gab es aber auch eine ganze Reihe weitere Familien, die ich nach dem Lesen der mit zugeschickten Befunde ebenfalls zur Einholung einer Zweitmeinung in einer anderen Uni-Klinik ermutigen konnte, in deren Folge sich die ihnen anfangs mitgeteilte Diagnose eines schweren MCAD-Mangels dann immerhin zu einer eindeutig milden und bisher nie in Form einer Entgleisung in Erscheinung getretenen Variante abschwächte. Bei der überwiegenden Mehrheit sprachen die Befunde wirklich ganz deutlich für einen schweren MCAD-Mangel, da diese Form tatsächlich auch die am häufigsten anzutreffende Variante ist. Die insgesamt ca. 50 Fälle der im Nachhinein dann doch nur milden Varianten oder reinen Carrier, zeigen aber bereits deutlich genug, wie wichtig es ist, sich immer alle Befunde in Kopie aushändigen zu lassen und zu überprüfen, ob die den Eltern gegenüber gemachten Aussagen auch mit den vorliegenden Befundwerten in Einklang gebracht werden können.
Ansonsten gibt es in den Befunden natürlich auch einige typische Begriffe, die dir in Verbindung mit den unter der Frage “Auf welche Variante des MCAD-Mangels weisen die Screeningergebnisse hin?” zu findenden Erklärungen eine Vorstellung ermöglichen sollten.
Eindeutige Hinweise auf eine schwere Form liegen vor, wenn folgende Begriffe auftauchen:
Deutliche Hinweise auf eine milde Form liegen dagegen vor, wenn die Mutation c.199t>c (Y67H)
gefunden wurde.
Alle anderen Mutationskombinationen sind im Vergleich dazu so selten, dass es keine ausreichende Datenbasis gibt, die eine auf den Erfahrungen betroffener Patienten aufbauende Klassifizierung in schwer oder mild ermöglicht. Eine MCAD-Mangel-Ausprägung kann allerdings schwerlich als mild im Sinne von “gutartig” bezeichnet werden, wenn eine Person mit dieser Mutationskombination schon mal eine Stoffwechselentgleisung erlitten hat. Anhaltspunkte zur Einstufung einiger der rund 70 bekannten Mutationen sind im Artikel “Infos zu den Mutationen” zu finden.
Am ehesten lassen sich zur Klassifizierung der individuellen Ausprägung eines MCAD-Mangels noch die reinen Acylcarnitinwerte (C-Werte, vor allem C8) und deren Quotienten aus den Screenings heranziehen, auch wenn diese selbst nur einen Verdacht begründeten und noch keine endgültige Diagnose darstellten. Trotzdem sind das die deutlichsten Werte, da sich die Neugeborenen zum Zeitpunkt dieser ersten Blutprobenentnahme normalerweise in einer stark katabolen Stoffwechsellage befanden. Wenn die in der Antwort zu “Auf welche Variante des MCAD-Mangels weisen die Screeningergebnisse hin?” behandelten Werte eines Kindes deutlich auf eine schwere Form hinweisen, sind die gefundenen Mutationen irrelevant, und eine eventuell durchgeführte Restaktivitätsanalyse des Enzyms wird diese Einschätzung mit hoher Wahrscheinlichkeit nur bestätigen, statt sie zu widerlegen. Lediglich in den Graubereichen, in denen die Acylcarnitinwerte eine klare Eingruppierung in Carrier<->milde Form<->schwere Form nicht direkt zulassen, können diese Untersuchungen ein kleines Stück mehr Klarheit verschaffen.
Hin und wieder kommt es allerdings auch vor, dass die Genanalyse nur eine Mutation hervorbringt, die Screeningwerte zuvor aber sehr deutlich auf einen MCAD-Mangel (egal ob mild oder schwer) hingewiesen haben. Dies ist nicht als Hinweis darauf zu verstehen, dass selbst Carrier, die nur von einem Elternteil einen MCAD-Gendefekt geerbt haben, einen MCAD-Mangel ausprägen können, sondern dass bei dem betreffenden Kind − und dem anderen Elternteil − eine mit der angewendeten Analysemethode noch nicht ermittelbare zweite Mutation vorliegt, die in Kombination mit der bereits gefundenen Mutation diese nachweisbare Einschränkung des MCAD-Enzyms ausgelöst hat. Zum Beispiel können mit der Methode zur Ermittlung von “Punktmutationen”, also einzelner ausgetauschter Basen im DNA-Strang, keine “Deletions”, also Löschungen größerer DNA-Teilstränge gefunden werden. Umgekehrt taugt die Methode zur Ermittlung von Deletions nicht zum Auffinden von Punktmutationen. Oftmals beschränken sich die Labore bei der Suche nach den auslösenden Mutationen jedoch auf die erste Methode, sodass eine vom zweiten Elternteil eventuell beigesteuerte andere Mutationsart schlicht und einfach nicht gefunden werden kann, obwohl sie aufgrund der Werte des Kindes zwingend existieren muss.
Haben die Screeningwerte bereits den Verdacht auf einen reinen Carrierstatus nahegelegt? Dann kann das Auffinden einer einzelnen heterozygoten Mutation als Beweis dafür angesehen werden. Bei dagegen deutlich erhöhten Acylcarnitinwerten hat das Auffinden von nur einer Mutation jedoch keine entsprechende Bedeutung, denn es muss dann zwingend eine zweite Mutation vom anderen Elternteil geben, die nur nicht gefunden werden konnte.![]()
Als mein Sohn geboren wurde, gab es zwar auch schon eine in den Niederlanden entwickelte Methode zur Ermittlung der verbleibenden MCAD-Enzymaktivität, aber diese Methode konnte aufgrund ihrer Ungenauigkeit nur vage Anhaltspunkte, jedoch keine sichere Differenzierung zwischen den Bereichen “kein MCAD-Mangel / milder MCAD-Mangel / schwerer MCAD-Mangel” liefern.
Insofern gab es damals (2007) genaugenommen nur die Mutationsbestimmung als etablierte Methode (siehe “Die Diagnose des MCAD-Mangels“), um einen sicheren Nachweis des MCAD-Mangels zu erbringen und darüber hinaus vielleicht auch noch eine Aussage über dessen Schweregrad zu ermöglichen. Es war eigentlich ganz simpel: Falls die häufigste Mutation (K329E) in doppelter Ausprägung oder in Kombination mit einer anderen Mutation gefunden wurde, die zuvor schon mal im Rahmen einer erfolgten Entgleisung entdeckt worden war, musste es sich zwangsläufig um einen schweren MCAD-Mangel handeln, weil aus beiden Genkopien nur hochgradig beschädigte Enzyme gebildet werden konnten. Umgekehrt ließ sich aus dem Auffinden von bislang nicht in Erscheinung getretenen Mutationen (auch in Kombination mit einer als schwer bekannten Mutation auf der anderen Genkopie) die Vermutung ableiten, dass es sich um einen Defekt handeln könnte, der das daraus gebildete Enzym nur unwesentlich beeinträchtigt. Einigermaßen sicher konnte man sich diesbezüglich aber nur bei Mutationen sein, die bereits ziemlich häufig gefunden wurden.
Bei allen anderen Kombinationen mit selteneren Mutationen hatten die Eltern dann zwar die Mutationsbezeichnungen auf dem Befundzettel stehen, konnten damit aber größtenteils nichts anfangen. Was nützt es einem schon, wenn man die Kombination K329E / A56P mitgeteilt bekommt, und es dann seitens der Klinik heißt: “Darüber gibt es bislang keine Informationen, also gehen wir einfach mal von einer schweren Ausprägung des MCAD-Mangels aus.” Das ist natürlich die für die Sicherheit des Kindes richtige Betrachtungsweise, aber genaugenommen hat die Untersuchung und das oft sehr lange Warten auf das Ergebnis dann letztlich zu keinem für die Eltern nützlichen Informationsgewinn geführt.
Tatsächlich haben in vielen Fällen bereits die Werte des Neugeborenenscreenings eine deutlichere Sprache hinsichtlich der Schwere des MCAD-Mangels gesprochen, als die später ermittelten Mutationsbezeichnungen. Ich habe im Laufe der Jahre sehr vielen Forumsteilnehmern, die mir ihre Screeningwerte mit der Bitte um eine Einschätzung geschickt haben, meist schon anhand dieser Werte prognostizieren können, welche Mutationskombination mit großer Wahrscheinlichkeit bei ihrem Kind gefunden werden würde und ob es sich um eine schwere oder milde Ausprägung handelte, bzw. dass sie selbst beim Auffinden einer eventuell abweichenden Kombination unbedingt von einem schweren MCAD-Mangel ausgehen müssten, weil die Screeningwerte keinen anderen Schluss zuließen.
Obwohl diese Einschätzungen so gut wie immer von dem Ergebnis der Mutationsbestimmung voll bestätigt wurden, möchte man als Eltern eines betroffenen Kindes speziell das Vorliegen eines milden und somit möglicherweise harmlosen MCAD-Mangels auch gerne eindeutig von ärztlicher Seite bestätigt bekommen, und dazu nützt es dann leider sehr wenig, wenn in dem Befund ein oder zwei sehr seltene oder gar völlig unbekannte Mutationen genannt werden, zu deren Auswirkungen es bislang keine gesicherten Erkenntnisse gibt.
Aus diesem Grund halte ich die heutzutage viel genauere und sehr verlässliche Ergebnisse liefernde Residualaktivitäts-Messung für die definitiv bessere Untersuchungsalternative. Indem damit direkt nachgewiesen wird, wieviel die fehlerhaft gebildeten MCAD-Enzyme tatsächlich noch zu leisten in der Lage sind, kann ein Prozentanteil einer “normalen” Enzymaktivität ermittelt werden, anhand dessen sich abschätzen lässt, ob die betreffende Person beim Vorliegen von “metabolischem Stress” aufgrund eines Magen-Darm-Infekts, bei langen Nahrungspausen oder bei deutlich erhöhtem Energiebedarf noch ausreichend Energie aus den Fettsäuren gewinnen kann, oder ob es kritisch werden könnte.
Auch bei diesen ermittelten Prozentwerten gibt es einen mittleren Graubereich, bei dem man zur Sicherheit des Kindes die gleiche Vorsicht und Behandlung wie bei der klassischen, schweren MCAD-Variante aufwenden sollte, aber die Unsicherheit basiert dann nicht darauf, dass man über die bei dem Kind vorliegende Mutation einfach nur noch nichts weiß.
Wird nach der bereits erfolgten Residualaktivitäts-Messung auch noch eine zusätzliche Mutationsbestimmung angefragt, würde ich persönlich dies heutzutage vor allem davon abhängig machen, ob diese Untersuchung kostenlos angeboten wird (zumindest früher machten das manche Einrichtungen noch für lau, um einfach nur Daten für die eigenen Forschungsarbeiten generieren zu können), oder wie hoch die ansonsten dafür veranschlagten Kosten sein sollen. Ich selbst sehe eigentlich keinen Sinn darin, für sehr viel Geld (ca. 2000€ für die molekulargenetische Untersuchung des Blutes des Kindes) auch noch die kryptischen Namen der verantwortlichen Mutationen zu erfahren, wenn die bereits erfolgten Untersuchungen schon deutlich den anzunehmenden Schweregrad des MCAD-Mangels aufgezeigt haben. Wie oben beschrieben bringt die Kenntnis um die Mutationsbezeichnung für die Eltern keinen darüber hinausgehenden Mehrwert, sondern nur hohe Kosten (für die Eltern selbst oder für die Krankenkasse − also uns alle), auf die man genauso gut verzichten kann.
Falls euch in eurer Stoffwechselambulanz nur die Mutationsbestimmung und nicht die Residualaktivitäts-Messung vorgeschlagen wurde, schreibt mich doch einfach mal per Email an − ich kann euch dann einen Tipp geben, an welche Stoffwechselklinik ihr euch diesbezüglich am besten wenden solltet.![]()
Ja, mit gewissen Abweichungen nach oben und unten bedingt jede einzelne Mutation des ACADM-Gens eine bestimmte Residualaktivität der aus dieser Genkopie (Allel) gebildeten MCAD-Enzyme. Besonders deutlich wird das bei den bekannten schweren Genmutationen, die zu einer fast vollständigen Funktionseinbuße der betreffenden Enzyme führen. Hier spielen sich die aus verschiedenen Blutproben ermittelten Aktivitäten in einem sehr engen Bereich ab.
Betrachten wir aber zuerst die für einen Menschen ohne MCAD-Mutationen angenommene “normale” Enzymaktivität von 100%. Sind keine Mutationen in den von den Eltern geerbten MCAD-Genen enthalten, bezeichnet man diese beiden Allele als “Wildtyp” oder kurz WT. Diese beiden WT-Allele produzieren MCAD-Enzyme mit einer messbaren Aktivität von insgesamt rund 100%. Natürlich ist dieser Wert nicht einfach so vom Himmel gefallen! Zur Festlegung, welche Enzymaktivität als “normal” und somit als 100%-Marke angesetzt werden kann, wurden von einer größeren Gruppe von Testpersonen, mit einwandfrei genetisch nachgewiesenen zwei WT-Allelen, die Residualaktivitäten bestimmt und der sich aus allen Ergebnissen ergebende Mittelwert als 100%, also als durchschnittliche, normale MCAD-Enzymaktivität festgelegt. Da sich diese 100% als Summe der Aktivitäten der von der Mutter und vom Vater geerbten WT-Allele darstellen, trägt jedes einzelne dieser beiden Allele somit eine Aktivität von bis zu 50% zu diesem Gesamtergebnis bei – weiter unten etwas mehr dazu, dass das längst nicht immer der Fall ist. Wenn nur eines der beiden Allele ein WT ist und das andere eine Mutation X enthält, kann man also aus einer ermittelten Residualaktivität von z.B. 52% ableiten, dass die 50% vom WT beigesteuert wurden und die andere Mutation X gerade mal 2% Restaktivität aufweist. Wenn nun diese Mutation X mit noch einer anderen Mutation Y und einer Residualaktivität von 10% gefunden wird, lässt sich aufgrund der bereits bekannten ca. 2% für Mutation X nun weiter berechnen, dass die Mutation Y dann mit ungefähr 8% angenommen werden kann, usw.
Zum weiteren Verständnis ist es also wichtig zu wissen, dass sich die Aktivitäten der von den beiden Allelen gebildeten Enzyme immer als Summe beider Varianten darstellen.
So wurden für die am häufigsten auftretende Mutationskombination c.985a>g (K329E) homozygot Residualaktivitäten von etwa 0,1% bis 0,9% gemessen, also im Mittel knapp 0,5%. Da bei den betreffenden Menschen zwei Allele mit der K329E-Mutation vorliegen, produziert somit jedes einzelne dieser beiden Allele MCAD-Enzyme mit einer Residualaktivität von genau der Hälfte, also im Mittel nur 0,25%. In gleicher Weise lässt sich auch bei anderen homozygot vorliegenden Mutationen nach dem Ermitteln der Residualaktivität sehr leicht berechnen, welche anteilige Restaktivität diese Mutation bei heterozygotem oder compound heterozygotem Auftreten zur Gesamtaktivität beiträgt. In entsprechenden Studien wurde dann aus einer großen Anzahl vorliegender Daten mit Mutationskombinationen und zugehörigen Residualaktivitäten, nach und nach ermittelt, welche Aktivitäten den einzelnen Allelen zugeschrieben werden können. Das ist nicht besonders kompliziert, sondern mit der Lösung einer mathematischen Gleichung a + b = c vergleichbar. a und b sind die zu zwei unterschiedlichen Mutationen gehörenden Einzelaktivitäten und c die bei dem Patienten mit dieser Mutationskombination gemessene Gesamtaktivität. Sobald der Wert von Aktivität a oder b bekannt ist, kann der noch unbekannte zweite Wert ganz einfach berechnet werden. Mehr ist es nicht. Am Ende erhält man eine stetig wachsende Tabelle für die von den unterschiedlichen Mutationen, also Allel-Varianten verantworteten Einzelaktivitäten.
Stellt man z.B. bei einer neuen, also hinsichtlich ihrer Auswirkungen noch nicht näher beschriebenen Mutationskombination K329E/Y220I eine Residualaktivität von 10% fest, kann aufgrund des bereits bekannten Beitrags der K329E von im Mittel 0,25% berechnet werden, dass die noch unbekannte Y220I ihrerseits die restlichen 9,75% beigesteuert hat. Das ist dann zwar schon mal deutlich mehr, als der Anteil der K329E, aber immer noch ein Bereich, in dem eine Mutation als “severe”, also schwerwiegend klassifiziert wird. Ebenso verhält es sich bei der Mutationskombination Y67H/K239N mit einer Gesamtaktivität von 18%. Aus bereits vielen Untersuchungen der relativ häufigen Y67H (c.199T>C) weiß man, dass Enzyme mit dieser Mutation für sich alleine eine Aktivität zwischen 16% und 21% aufweisen. Insofern kann der Anteil der Enzyme mit der K239N-Mutation auch nur im Bereich von nix bis knapp über 0 liegen. Damit ist diese Mutation eindeutig als schwer zu klassifizieren.
In der erwähnten Studie, mit einem Vergleich sehr vieler unterschiedlicher Mutationskombinationen und der zugehörigen Residualaktivitäten, hat sich gezeigt, dass es unter den von den einzelnen Mutationen verursachten Restaktivitäten so ziemlich alle Abstufungen von 0% bis ungefähr 34% gibt. So wurde für die Kombination K329E/A218N eine Aktivität von 34% gemessen. Angesichts des Umstandes, dass die K329E zu diesen 34% im besten Fall 0,5% beigetragen hat, kommen also fast die gesamten 34% dieser Kombi von der A218N. Damit ist ihre Residualaktivität fast doppelt so hoch, wie die der schon lange als mild angenommenen Y67H.
Wenn du dir die Diagramme der Studie anschaust, könntest du an einer bestimmten Stelle stutzig werden: Es gibt da eine ganze Reihe Kombinationen von Mutationen mit einem WT-Allel, die in ihrer Gesamtaktivität deutlich unter 50% liegen, in einem besonders gravierenden Fall sogar nur bei 25%. Moment mal! Wenn ein Wildtyp-Allel zu Bildung von vollkommen normal funktionierenden MCAD-Enzymen führt und die mutierten Allele zusätzlich immer noch einen kleinen Teil zusätzlich dazu beitragen, dann müssten doch alle Personen mit WT-Anteil (also alle Carrier) automatisch mindestens mal 50% oder sogar noch deutlich darüber haben, oder?
Das trifft nur in der Theorie zu, in der Praxis nicht, denn es gibt eine ganze Reihe von Umständen, die dazu führen können, dass die aus einem Wildtyp-Allel erzeugten MCAD-Enzyme nicht die volle Leistung bringen oder zu bringen scheinen. Zumindest mal in den Blutproben, die zur Residualaktivitätsanalyse eingesendet werden und dafür eine unter Umständen mehrtägige und “strapaziöse” Reise auf sich nehmen müssen. Eine gewisse Schädigung der Probe kann also schon durch einen sehr langen oder nicht optimal gekühlten Transport passieren, sodass die daraus “gezüchteten” Enzyme nicht in vollem Umfang analysiert werden können. Bevor du jetzt aber denkst, dass die bei deinem Kind auf einen schweren MCAD-Mangel hindeutende Residualaktivität vielleicht einfach nur aufgrund einer beschädigten Blutprobe zustande kam und es daher in Wirklichkeit vielleicht nur einen milden oder sogar gar keinen MCAD-Mangel habe, muss ich diesen Gedankengang gleich bremsen. In den Analyse-Laboratorien kann man schon ziemlich genau erkennen, ob eine eingesandte Blutprobe noch aussagekräftige Ergebnisse liefern kann, oder verdorben und somit unnütz ist.
Darüber hinaus wurden für diese Studie über einen Zeitraum von sechs Jahren eingesandte Blutproben aus Deutschland, Kanada und Italien ausgewertet – allesamt von Neugeborenen, die im NG-Screening hinsichtlich eines MCAD-Mangels auffällig waren. Es handelt sich also trotz der vermeintlich großen Testgruppe um einen nur verschwindend geringen Anteil aller Kinder, die im gleichen Zeitraum in diesen drei Ländern geboren wurden, und von denen fast alle zwei WT-Allele haben und ein kleiner, aber immer noch weitaus größerer Anteil als die in der Studie aufgeführten rund 50 Kinder, die ein WT-Allel und eine andere Mutation aufweisen. Über diese vielen Millionen anderer Kinder kann man keine Aussage treffen, sondern nur für die paar wenigen Kinder, für die in diesem Zeitraum eine Blutprobe mit einem Transportweg von teilweise mehreren Tagen eingesendet wurde. Die 256 Proben umfassende Kontrollgruppe mit zwei intakten WT-Allelen diente nur zur Bestimmung eines durchschnittlichen 100% Standard-Wertes, hat aber vermutlich keine gleichermaßen weiten Wege zurücklegen müssen. Aus dieser Kontrollgruppe wird jedoch ganz deutlich, dass man einem einzelnen WT-Allel im Normalfall einen Beitrag von 50% zuschreiben kann, der halt von den anderen eingesendeten Proben zum Teil nicht mehr ganz erreicht werden kann.
Für die eine Probe, bei der die Residualaktivität trotz des enthaltenen WT-Allels sogar nur bei 25% liegt, wird seitens der Verantwortlichen für die Studie vermutet, dass in Wirklichkeit doch eine Mutation auf dem vermeintlichen WT-Allel vorliegen dürfte, die mit den zur Mutationssuche herangezogenen Verfahren nicht gefunden werden konnte. Es kann z.B. vorkommen, dass bei der Suche nach einzelnen Punktmutationen nichts gefunden wird, weil ein größerer Abschnitt der DNA aufgrund einer Deletion einfach nicht da ist. Wo nichts ist, werden keine Fehler gefunden, aber trotzdem ist das Gen und somit das daraus gebildete Enzym hochgradig geschädigt. Ob ein ähnliches Problem in diesem Fall vorlag, ist nicht beschrieben, aber der niedrige Aktivitätswert legt nahe, dass dieser eine WT halt doch kein echter WT ist.
Der wichtigste Grund aber, weshalb Kombinationen von Mutation mit WT-Allel oftmals unterhalb von 50% liegen, liegt darin, wie der zum Vergleich angenommene Referenzwert von 100% bestimmt wird. Diese 100% sind immer der Mittelwert aus sehr vielen völlig individuellen Einzelaktivitäten. Um es mit einem anderen Bild auszudrücken: bezogen auf den IQ der Menschen in Deutschland kann man mit Recht sagen, dass die Hälfte aller in Deutschland lebenden Menschen unterdurchschnittlich intelligent ist. Das ist nicht beleidigend gemeint, sondern ergibt sich zwingend aus der Festlegung der 100%-Marke für den “normalen” IQ. Gleichzeitig ergibt sich daraus nämlich auch, dass die andere Hälfte der Menschen in Deutschland überdurchschnittlich intelligent ist. Das liegt einfach nur daran, dass die offiziellen, standardisierten IQ-Tests an einer sehr großen Zahl von Menschen ausprobiert wurden und dann die 100er-Marke für die “durchschnittliche” Intelligenz so gelegt wurde, dass genau 50% der Testpersonen schlechter und 50% besser abgeschnitten haben.
Bei der Berechnung der durchschnittlichen “normalen” Residualaktivität der Testpersonen mit zwei WT-Allelen verhält es sich ähnlich. Zur Bestimmung des Mittelwertes können z.B. die Enzymaktivitäten von 199 Testpersonen zusammengezählt und durch 199 geteilt werden, oder man sortiert diese 199 Einzelergebnisse der Größe nach und nimmt als Referenzwert einfach den dann genau in der Mitte liegenden Wert an Position 100. Dann sind 99 Werte kleiner und 99 größer. Das ist der sogenannte Median. Aber wie auch immer der für solche Residualaktivitäten verwendete Referenzwert von 100% bestimmt wird – es gibt unter den dafür herangezogenen Einzelaktivitäten auch deutliche Ausreisser nach unten und oben. So gibt es unter Menschen mit zwei WT-Allelen also sowohl solche, die deutlich unter dem Referenzwert von 100% liegen, als auch solche, die deutlich darüber liegen.
Bei der Bestimmung der Residualaktivität einer einzelnen Person mit einer bestimmten Kombination aus Mutation und WT-Allel, wird aber immer deren individuelle WT-Aktivität mit ins Gewicht fallen, die dann in den seltensten Fällen genau 50% entspricht. Dadurch wird die Berechnung des Aktivitäts-Anteils der bei diesem Menschen dann nur heterozygot vorliegenden Mutation deutlich erschwert.
Beispiel 1: Bei der Untersuchung von vier Menschen mit der Kombination WT/c.199T>C wird eine durchschnittliche Residualaktivität von 70% (bezogen auf den 100%-Referenzwert) festgestellt. Die Einzelergebnisse der vier Testpersonen reichen aber von 60% bis 80%. Bringt die c.199T>C also bei dem einen Patienten tatsächlich nur 10% zusätzlich zu den 50% des WT-Allels und bei der anderen Testperson dagegen sogar 30%? Glücklicherweise handelt es sich bei der c.199T>C auch um eine sogenannte “prävalente”, also vorherrschende Mutation, die bei den im Neugeborenenscreening auffälligen Kindern dann doch so häufig auftaucht, dass man noch andere Vergleiche ziehen kann. Im Zusammenspiel mit der c.985A>G (K329E) werden üblicherweise Werte von um die 20% ermittelt. Da die K329E dazu so gut wie nichts beiträgt, kann man aus einer somit deutlich größeren Datenbasis ableiten, dass die c.199T>C eine relativ konstante Aktivität von ca 20% beisteuert und somit der WT-Anteil bei den dafür heterozygoten vier Testpersonen also nicht genau bei 50% liegen wird, sondern vermutlich von 40-60% reichen muss, so dass insgesamt dann die Bandbreite von 60-80% mit dem Mittelwert 70% herausgekommen ist.
Beispiel 2: Bei der ebenfalls als sehr mild und daher vermutlich nicht krankheitsrelevant angenommenen c.127G>A (E47K) kommt in Kombination mit K329E bei drei Patienten sogar eine mittlere Residualaktivität von 31% (26-36%) heraus, also sogar noch einige Prozentpunkte höher als die c.199T>C. In der Vergleichsgruppe der heterozygoten Carrier für diese c.127G>A Mutation liegt die Residualaktivität allerdings nur bei 65%, also deutlich unter dem Ergebnis der c.199T>C. Wie kann das sein? Hier muss berücksichtigt werden, dass diese 65% nur von einer einzigen heterozygoten Testperson stammen und sich somit deren individuelle Abweichung des Beitrags des WT-Allels voll auf dieses eine Ergebnis auswirkt. Hätte man Vergleichsdaten von noch einigen anderen Carriern dieser Mutation, würde sich dieser eine vergleichsweise niedrige Wert von 65% durch die Hinzuziehung der anderen teilweise deutlich höheren Aktivitätswerte mehr und mehr zu einem passenderen Mittelwert hin korrigieren.
Aus dem gemeinsamen Vorkommen einer bestimmten Mutation zusammen mit der K329E kann man also deren eigenen Aktivätsanteil viel deutlicher ableiten, als aus der Kombination mit einem WT-Allel, dessen Eigenaktivität sowohl ein ganzes Stück unter als auch über den angenommenen 50% liegen kann. Bei der Betrachtung von Tabellen oder Diagrammen (wie z.B. in der zuvor erwähnten Studie) mit Mutationskombinationen und deren Residualaktivitäten, muss man ganz allgemein im Blick behalten, dass es sich bei nur auf einzelnen Personen basierenden Testergebnissen um deutliche Ausreisser handeln kann und die Einschätzung der tatsächlichen mittleren Restaktivität einer bestimmten Mutation umso deutlicher und verlässlicher wird, je mehr Ergebnisse dafür herangezogen werden können.
Solltest du zu den Eltern gehören, deren Kind leider schon kurz nach der Geburt − meist schon vor dem Vorliegen des auf einen MCAD-Mangel hindeutenden Screening-Ergebnisses − eine erste Stoffwechselentgleisung erlitten hat, wurde dir vielleicht von einem Arzt mitgeteilt, dass ausgerechnet dein Kind an einer anscheinend ganz besonders extrem schweren Form des MCAD-Mangels zu leiden scheint. Mit dieser dann doch leider sehr unbedachten Äusserung eines davon völlig überraschten und sich meistens auch nicht sonderlich gut mit dem MCAD-Mangel auskennenden Arztes, wird den Eltern zusätzlich zu dem bereits erlittenen Schock auch noch in Aussicht gestellt, dass ihr Kind noch viel stärker als alle anderen in “üblicher Weise” vom MCAD-Mangel betroffenen Kinder gefährdet sei, weitere gefährliche Entgleisungen zu erleiden. Tatsächlich ist diese Behauptung aber an den Haaren herbeigezogen und beruht lediglich auf mangelnder Erfahrung des betreffenden Arztes hinsichtlich der verschiedenen Ausprägungen des MCAD-Mangels − wenn er denn überhaupt schon mal mit einem davon betroffenen Kind zu tun hatte. Wenn der Arzt nicht gerade Experte für den Schwerpunkt Stoffwechselstörungen bei Kindern ist, hat er mit sehr großer Wahrscheinlichkeit ohnehin noch nie zuvor vom MCAD-Mangel gehört, nur mal schnell ein paar Zeilen nachgelesen und dabei erfahren, dass schwere Stoffwechselkrisen früher zwar auch schon in den ersten paar Lebensjahren, aber selten so kurz nach der Geburt eingetreten sind (hängt auch damit zusammen, dass die Mütter früher auf den Säuglingsstationen sehr viel schneller und intensiver zum Zufüttern gedrängt wurden, um ein schreiendes Kind zur Ruhe zu bringen, wenn der Milcheinschuss noch etwas auf sich warten ließ und man heute mehr und mehr davon abkommt). Was läge also näher, als anzunehmen, dass es sich bei dem vorliegenden Fall somit um eine ganz herausragend schwere Störung der betreffenden Stoffwechselfunktion handeln muss − sonst wäre ja schließlich nichts passiert, oder?
Unter einer “normalen”, also schweren Form des MCAD-Mangels, versteht man die Ausprägungen, bei denen die beiden von den Eltern an das Kind vererbten MCAD-Gene (Allele) einen so gravierenden Schaden aufweisen, dass der in ihnen enthaltene Bauplan für die Synthese des MCAD-Enzyms in beiden Kopien dermaßen stark beschädigt ist, dass entweder ein unvollständiges oder durch Fehlfaltungen funktionsunfähiges Enzym gebildet wird. Egal anhand welcher der beiden Genschablonen ein MCAD-Enzym zusammengebaut wird, es kommt immer nur Murks dabei raus. Insofern können beide bei dem Kind existierenden Formen von MCAD-Enzymen ihre Aufgabe der Aufspaltung der mittelkettigen Fettsäuren nicht erledigen und der Vorgang bricht an dieser Stelle ab. Welche Mutationen dabei an das Kind vererbt wurden und für diesen schweren Genschaden verantwortlich sind, ist dabei unerheblich. Letztlich läuft es immer auf das Gleiche hinaus − die MCAD-Enzyme haben keine Funktion und somit auch keine Restaktivität. Sobald diese Enzyme in der Verarbeitungskette zur Aufspaltung der Fettsäuren an die Reihe kommen, bricht dieser Vorgang ab, da die Enzyme ihre Arbeit nicht tun können.
Die am häufigsten, also bei den meisten Kindern mit MCAD-Mangel anzutreffende Mutationskombination K329E homozygot fällt genau in diese Kategorie. Es spielt dann auch keine Rolle, ob bei der Messung der Residualaktivität bei einem Kind glatte 0% und bei einem anderen Kind zufällig 1% oder 2% herauskommen. Wenn beide Kinder die schwere Variante K329E homozygot aufweisen, hat das Kind mit 2% keineswegs eine leichtere schwere Variante und das Kind mit 0% auch nicht eine noch schwerere schwere Variante. Während es bei einer Geldanlage über die Jahre hinweg sehr wohl einen großen Unterschied macht, ob der angelegte Betrag mit 0%, 1% oder 2% verzinst wird, sind hinsichtlich der Restaktivität eines Enzyms in diesem Wertebereich die geringen Unterschiede in den Prozenten völlig belanglos. Auch das ist eines der Themen, bei denen im Gespräch unter MCAD-Eltern keine Werte verglichen und diskutiert werden sollten.
Ich habe gerade in den letzten Jahren des früheren Forums, als zur Diagnose des MCAD-Mangels immer häufiger die Residualaktivitätsanalyse herangezogen wurde, schon viele Male erlebt, dass sich neue Teilnehmer mit dem Informationszusatz vorstellten “… und mein Kind hat einen schweren MCAD-Mangel mit 2% Restaktivität.” Worauf sich andere, ähnlich neue Teilnehmer veranlasst sahen, mit einem “bei meinem Kind sind es glatte 0% Restaktivität” zu antworten, woraufhin nicht selten eine Diskussion entstand, ob die 2% dann nicht doch deutlich besser oder die 0% noch wesentlich schlechter seien. Um dies mal mit einem ganz anderen Beispiel zu verdeutlichen: Wenn man einen kleinen Hunger verspürt, findet man in einem Supermarkt für 1€, also 100 Cent, zwar nicht viele, aber doch noch eine kleine Auswahl an Lebensmitteln, unter denen man wählen kann, um seinen Hunger zu stillen. Für 25 Cent findet man in den meisten Läden (vor allem den Discountern) immerhin noch eine billige Sorte Brötchen, die den gleichen Effekt haben. Aber ob man nun noch 2, 3 oder 4 Cent einstecken hat oder gar kein Geld mehr, ist völlig egal, denn dafür bekommt man heutzutage in keinem Laden mehr irgendetwas. Genauso verhält es sich mit den Residualaktivitäten. Ob es 0%, 1% oder auch mal 2% sind, ist auch von der jeweiligen Tagesform abhängig oder die Untersuchungen wurden einfach nur in unterschiedlichen Kliniken durchgeführt, so dass leicht voneinander abweichende Referenzwerte zugrunde gelegt wurden. Würde die Untersuchung mehrfach wiederholt, könnten bei einem Kind mit zuerst 1% in der zweiten Runde auch 0% rauskommen oder umgekehrt. In anderen Wertebereichen, z.B. für die vermutlich milden MCAD-Varianten, verhält es sich ebenfalls so. Ob bei einem Kind 23% Residualaktivität oder 25% gemessen werden, ist ebenfalls unwichtig. Es geht nur darum, dass das bei dieser einen Untersuchung ermittelte Ergebnis eindeutig in einem Bereich liegt, von dem man annehmen kann, dass die betreffenden Kinder in schwierigen Krankheitssituationen − den bisherigen Erfahrungen zufolge − nicht in eine gefährliche Stoffwechselkrise gleiten werden.
Kleiner Einschub: die gerade genannten 25% hören sich immer noch nach wenig an, aber man muss dabei bedenken, dass im Körper eines Lebewesens Stoffwechselvorgänge immer in Regelkreisen gesteuert werden. Es gibt nicht eine feste Anzahl von Enzymen für die Fettsäuren mit bestimmten Längen, sondern sie werden ununterbrochen – je nach Bedarf – aus Aminosäurenketten erzeugt und auch wieder abgebaut. Wenn nach der Verkürzung auf mittelkettige Restlänge mehr dieser Fettsäuren in den Mitochondrien rumschwirren, als in kurzer Zeit von den momentan vorhandenen MCAD-Enzymen verarbeitet werden können, werden einfach noch ein paar mehr der betreffenden Enzyme gebildet. Bei milden Varianten, bei denen also mindestens eine der beiden Genkopien noch MCAD-Enzyme mit einer gewissen Restfunktion definiert, gibt es dann durch die gesteigerte Enzymproduktion zwar auch mehr MCAD-Enzyme, die gar nichts können (weil sie anhand des Gens mit der schwereren Mutation gebildet wurden), aber auch deutlich mehr MCAD-Enzyme mit der noch zum Teil vorhandenen Restfunktionalität, die dann entsprechend mehr mittelkettige Fettsäuren weiter verkürzen können. Diese Regelkreise haben natürlich auch ihre Grenzen, denn bei zwei Genkopien mit schwerer Mutation nützt es auch nichts, einfach mehr MCAD-Enzyme zu produzieren. Selbst wenn ihre Anzahl verfünffacht oder verzehnfacht werden könnte – 10 mal nix ist immer noch nix. Aber gerade bei Menschen mit mildem MCAD-Mangel lässt sich dadurch erklären, warum es trotz der insgesamt ja doch beträchtlich reduzierten Enzymaktivität so gut wie nie zu irgendwelchen Symptomen kommt, wie sie in den gleichen Situationen beim schweren MCAD-Mangel eintreten würden. Und bei heterozygoten Carriern, deren eine nicht betroffene Genkopie vollkommen intakte MCAD-Enzyme definiert, werden durch die automatisch gesteigerte Enzymproduktion auch davon wieder so viele Exemplare erzeugt, dass die mittelkettigen Fettsäuren problemlos weiter bearbeitet werden können.
Dass es bei manchen Kindern mit schwerem MCAD-Mangel dann doch so kurz nach der Geburt zu einer ersten Entgleisung kam und bei anderen nicht, hängt einfach von den Begleitumständen ab. Das eine Kind ist vielleicht ein Erstgeborenes, sodass es mit dem Milcheinschuss bei der Mutter einfach noch ein paar Tage dauerte und es daher in den ersten zwei Tagen kaum Nahrung zu sich nehmen konnte − bei einem anderen Kind ging es mit dem Stillen schneller. Eine Mutter hat vielleicht dem vehementen Drängen der Säuglingsschwestern nachgegeben und nach kurzer Zeit Neugeborenennahrung zugefüttert, während eine andere Mutter dies abgelehnt und weiter versucht hat, voll zu stillen. Ein Kind hat vielleicht eine Neugeboreneninfektion bekommen, für deren Bekämpfung sein Körper noch viel mehr Energie aufwenden musste, als ein anderes Kind, das soweit völlig gesund war.
Letzten Endes ist immer nur das unglückliche Zusammenspiel der Begleitumstände dafür verantwortlich, weshalb es bei einem Kind mit schwerer MCAD-Variante so schnell zu einer Entgleisung kommt und bei einem anderen Kind mit genauso schwerer Ausprägung nicht. Es liegt jedenfalls nicht daran, dass es noch viel schlimmere schwere MCAD-Ausprägungen gäbe und Kinder mit bereits kurz nach der Geburt erlittener Entgleisung sind im weiteren Leben auch keineswegs stärker gefährdet, als Kinder mit gleich schwerer Ausprägung, denen dieses Erlebnis glücklicherweise erspart geblieben ist.
Ganz abgesehen von diesen unterschiedlichen Erfahrungen in frühester Kindheit, entwickeln sich auch die heranwachsenden Kinder völlig individuell. Manche Kinder überragen beim Schuleintritt ihre gleichaltrigen Klassenkameraden schon um eine ganze Kopfhöhe, hören aber vielleicht bereits mit 14 Jahren auf zu wachsen, während es bei den bis dahin immer kürzeren Mitschülern noch für ein paar Jahre weiter geht. Bei einigen Jungen prägt sich eine deutlich tiefere Stimme schon mit 12 Jahren innerhalb weniger Wochen aus, während andere über Jahre hinweg vor sich hin quietschen und sich wieder andere noch mit 14 Jahren nicht viel anders anhören, als zu Grundschulzeiten. Genausowenig, wie man also Kinder ohne MCAD-Mangel hinsichtlich ihrer Entwicklung miteinander vergleichen kann, kann man solche Vergleiche bei Kindern mit schwerem MCAD-Mangel anstellen. Im realen Leben spielen immer eine ganze Menge unterschiedlicher Faktoren eine Rolle, wenn es darum geht, wie heftig unterschiedliche Kinder von einer Krankheitssituation beeinträchtigt werden oder wie schnell sie sich hinsichtlich Körpergröße und körperlicher oder geistiger Reife entwickeln. Ein Kind (ohne MCAD-Mangel) steckt jede Erkältung mit ein paar Tagen verstopfter Nase, ansonsten aber weitestgehend körperlich fit weg, ein anderes Kind (ebenfalls ohne MCAD-Mangel), das sich bei dem ersten Kind mit dem gleichen Erreger angesteckt hat, liegt dagegen ein paar Tage flach und fühlt sich von der Erkältung völlig ausgelaugt. Oftmals spielen dabei auch weitere gesundheitliche Faktoren eine Rolle, denn auch wenn bei einem Kind frühzeitig ein MCAD-Mangel diagnostiziert wurde, bedeutet dies keineswegs, dass nicht noch irgendwelche anderen gesundheitlichen Beeinträchtigungen bei ihm vorliegen können, die nur noch nicht gefunden wurden. Insofern könnte man zwar tatsächlich behaupten (das machen auch manche Ärzte), dass auch kein schwerer MCAD-Mangel wie der andere ist − tatsächlich ist jedoch die hinter dem schweren MCAD-Mangel steckende Funktionsstörung bei allen davon betroffenen Kindern identisch (weniger als nichts geht nicht), aber durch die bei allen Kindern vollkommen individuellen Begleitumstände, können die im tagtäglichen Leben zu beobachtenden Auswirkung des gleichen schweren MCAD-Mangels ganz unterschiedlich und zum Teil halt auch viel deutlicher ausfallen. Im Artikel “Woher kommt der MCAD-Mangel” wird im Abschnitt “Aus dem Genotyp folgt nicht der Phänotyp” noch einmal auf diesen Umstand eingegangen.![]()
Diese Frage lässt sich schwer beantworten, denn die Gesamtanzahl aller vom MCAD-Mangel betroffenen Menschen in Deutschland lässt sich nur grob schätzen. Einige wurden frühzeitig anhand des Screenings gefunden, andere bekamen die MCAD-Diagnose erst nach einer Stoffwechselentgleisung oder zumindest gesundheitlichen Problemen, wieder andere leben seit vielen Jahren völlig ahnungslos mit ihrem MCAD-Mangel, weil sie bisher keine Auffälligkeiten entwickelt haben und somit auch nie diagnostiziert wurden.
Über den Anteil der vom MCAD-Mangel betroffenen Kinder, die inzwischen dank des erweiterten Neugeborenenscreenings frühzeitig gefunden werden, liefern die jährlichen Screeningberichte der Deutschen Gesellschaft für Neugeborenenscreening e.V. (DGNS) einen ersten Anhaltspunkt. Aus den auf der Webseite verfügbaren Berichten der Jahre 2004 bis 2019 geht hervor, dass in diesen 16 Jahren insgesamt rund 1100 Neugeborene mit MCAD-Mangel diagnostiziert werden konnten. Dabei handelt es sich um die nach dem Recall (Kontrollscreening) übrig gebliebenen positiven Fälle. Pro Jahr kommen somit durchschnittlich etwa 68 neue, bestätigte MCAD-Fälle hinzu, was sich in dieser Größenordnung auch für die Jahre 2020 bis heute fortsetzen dürfte.
Da das um den MCAD-Mangel und ein paar andere seltene Störungen erweiterte Neugeborenenscreening schon 1999 in Bayern als Modellprojekt startete, und sich erst nach und nach die Screeningzentren weiterer Bundesländer anschlossen, wurden in den Jahren vor 2004 noch nicht alle Neugeborenen gescreent und somit auch nur ein Teil der betroffenen Kinder frühzeitig gefunden. Einige der nicht gescreenten Kinder wurden aufgrund erlittener Stoffwechselkrisen im Nachhinein noch mit dem MCAD-Mangel diagnostiziert, viele verstarben jedoch auch infolge einer solchen Krise.
Man kann somit hochrechnen, dass es bis heute (Mitte 2023) etwa 1400 bis 1500 diagnostizierte MCAD-Betroffene in Deutschland geben dürfte. Da der MCAD-Mangel aber nicht erst seit 1999 existiert, sondern auch alle lebenden älteren Jahrgänge in gleicher Weise und mit vermutlich auch annähernd gleicher Häufigkeit davon betroffen sind, muss man bei der Frage, wie viele Menschen mit MCAD-Mangel insgesamt in Deutschland leben auch diejenigen berücksichtigen, die bisher nicht diagnostiziert wurden, und die somit auch überhaupt nichts von ihrer Betroffenheit ahnen. Diese Zahl ist nun wirklich nur noch ganz grob zu schätzen, denn je weiter zurück die Geburt der betroffenen Menschen liegt, desto größer ist die Wahrscheinlichkeit, dass es bei ihnen in der Vergangenheit irgendwann zu einer tödlich endenden Stoffwechselkrise kam. Auch wenn es schon in den letzten 90 Jahren pro Jahr bei 1 von 10000 Kindern zu einem MCAD-Mangel kam, wird die Anzahl der noch lebenden und nichts von ihrer Betroffenheit ahnenden Menschen mit steigendem Alter pro Jahrgang immer geringer werden. Meine persönliche Schätzung geht in die Richtung von derzeit vielleicht 2500 bis 3500 lebenden MCAD-Betroffenen in Deutschland, von denen aber nur die zuvor erwähnten 1400 bis 1500 diagnostizierten Patienten der letzten Jahre von ihrer Betroffenheit wissen.![]()
Ganz klar: NEIN! Ich persönlich würde den vielleicht bereits vereinbarten Termin für eine erneute Blutentnahme auch einfach absagen. Es sprechen nämlich zwei ganz wichtige Gründe dafür, weshalb bei deinem Kind kein milder MCAD-Mangel vorliegen kann:
Dass die Ärzte in der Klinik auch noch einen milden MCAD-Mangel ausschließen wollen (indem sie dazu nach mehreren Tagen und 3-4 Stunden Nahrungspause noch einmal Blut für ein erneutes Acylcarnitinprofil abnehmen), soll natürlich zu eurer, aber vor allem auch zu ihrer eigenen Sicherheit dienen, ist aber vollkommen unnötig. Wie oben gezeigt, kann dein Kind unter den vorliegenden Umständen überhaupt keinen milden MCAD-Mangel haben, sondern dieser weitere Abklärungs-Vorschlag (betrachte es ruhig nur als solchen!) resultiert daraus, dass man sich in der Klinik noch nicht sehr intensiv mit den Mechanismen des MCAD-Mangels und den dafür geltenden Vererbungsregeln auseinandergesetzt zu haben scheint. Vielleicht ist man sich auch bzgl. der Entstehung des milden MCAD-Mangels nicht so ganz klar und glaubt einfach nur, dass ein von beiden Elternteilen vererbter Gendefekt zu einem schweren MCAD-Mangel führt, und dass die Vererbung von nur einem defekten Gen dann einen milden darstellen würde. Das klingt im ersten Moment unwahrscheinlich, da man doch meinen sollte, dass sämtlichen Stoffwechselärzten bekannt sein müsste, dass ein MCAD-Mangel – egal ob schwer oder mild – zwingend einen beidseitigen Defekt, also auf jeder der beiden Genkopien voraussetzt. Es gab aber schon zu Forumszeiten genügend Eltern, denen gegenüber seitens ihrer Ärzte aus purer Ahnungslosigkeit heraus genau dies behauptet wurde: “Ihr Kind hat das Gleiche wie Sie, nur Sie haben es in einer milden Form und ihr Kind in einer schweren!”. Wer so etwas behauptet, setzt den reinen Carrier-Status mit einem milden MCAD-Mangel gleich und das ist Unsinn! Ein einzelnes defektes MCAD-Gen ist klinisch ohne Bedeutung und ist kein milder MCAD-Mangel. Es werden dann anhand der intakten Genkopie so viele funktionstüchtige Enzyme gebildet, dass die Fettsäurenoxidation ohne Probleme vollständig durchlaufen wird.
Mit einer großen Wahrscheinlichkeit von rund 67% ist dein Kind Anlagenträger, also Carrier für einen schweren MCAD-Mangel, weil es dazu von nur einem von euch beiden Elternteilen die entsprechende Anlage vererbt bekommen hat. Mit 33%iger Wahrscheinlichkeit hat es nur eure jeweils intakte MCAD-Genkopie bekommen und ist damit nicht mal Carrier. Die letzte Kombinationsmöglichkeit mit zwei vererbten Mutationen ist aufgrund des Screeningergebnisses bereits aus dem Rennen. Siehe dazu den Artikel “Woher kommt der MCAD-Mangel“.
Warum würde ich nun den Termin für eine weitere Blutentnahme absagen? Wie oben gezeigt, kann dein Kind keinen milden MCAD-Mangel haben und daher würde mit sehr großer Sicherheit auch das zweite Testergebnis unauffällig sein. Aus eigener Erfahrung weiß ich aber, dass unter ganz ungünstigen Umständen auch bei Carriern während der ersten paar Lebenstage (es dauert bis zu vier Wochen, bis der Stoffwechsel eines Neugeborenen richtig eingespielt ist) die im Blut gemessenen Acylcarnitinmengen mal vorübergehend minimal über den sehr niedrig gewählten Grenzwerten liegen können. Insgesamt unterscheidet sich das Acylcarnitinprofil (vor allem die Quotienten) eines solchen nur anfangs mal ganz leicht auffälligen Carriers sehr deutlich und in charakteristischer Weise von dem eines Neugeborenen mit schwerem oder mildem MCAD-Mangel, aber um das auf Anhieb zu erkennen, muss man schon ein wenig Ahnung mit vergleichbaren Fällen haben. Leider muss man aber eher damit rechnen, dass die sich damit nicht auskennenden Ärzte (so wie bei uns) unter diesen Umständen einfach mal wenigstens einen milden MCAD-Mangel als erwiesen ansehen würden, der von den meisten Stoffwechselkliniken dann auch nicht anders behandelt wird, wie ein schwerer. Es erfordert danach einiges an Aufwand und persönlichem Engagement von euch Eltern, um diese Diagnose mit Einholung einer Zweitmeinung zu widerlegen. Ihr habt mit dem unauffälligen Screeningergebnis aber bereits den eindeutigen und verlässlichen Nachweis, dass euer Neugeborenes keinen MCAD-Mangel hat und fertig! Lasst euch von niemandem mit wenig Ahnung einreden, dass da vielleicht doch etwas sein könnte. Ansonsten müssten auch die Eltern der anderen pro Jahr über 700.000 Neugeborenen, deren Screening hinsichtlich des MCAD-Mangels völlig unauffällig war, dieses Ergebnis in Zweifel ziehen. Unter diesen unauffälligen Neugeborenen gibt es schätzungsweise rund 12.000 Carrier. Trotzdem wird keines dieser Kinder noch einmal weiter in Richtung MCAD-Mangel untersucht. Der einzige Unterschied zu euch besteht lediglich darin, dass man für euer Neugeborenes aufgrund des MCAD-Mangels des Geschwisterkindes den Carrierstatus relativ sicher annehmen kann. Trotzdem besteht diesbezüglich kein Unterschied zwischen ihm und den 700.000 anderen mit unauffälligem Ergebnis gescreenten Kindern pro Jahr.![]()
Es ist richtig, dass man den MCAD-Mangel in eine gewisse Beziehung mit ein paar wenigen Fällen (ca. 1%-3%) von plötzlichem Kindstod bringen kann. Allerdings nur in der Form, dass man im Nachhinein − teilweise erst viele Jahre später − eine aufgrund eines unentdeckten MCAD-Mangels aufgetretene nächtliche Stoffwechselentgleisung, bzw. die sich in deren Folge einstellende Unterzuckerung und die daraus folgende Mangelversorgung des Gehirns und weiterer lebenswichtiger Organe als Todesursache identifizieren konnte.
Der plötzliche Kindstod (auch als Krippentod, Säuglingstod oder englisch als SIDS = sudden infant death syndrome bezeichnet) ist keine eigenständige Todesursache, sondern eine Ausschlussdiagnose. In der San Diego-Definition wird er beschrieben “als der plötzliche, unerwartete Tod eines Kindes, das jünger als ein Jahr ist. Das fatale Ereignis geschieht vermutlich während des Schlafes. Auch nach eingehender Untersuchung, einschließlich der Durchführung einer vollständigen Autopsie und der Begutachtung der Todesumstände sowie der medizinischen Vorgeschichte, bleibt der Tod ungeklärt.“ [Krous et al., 2004]. Die Diagnose “plötzlicher Kindstod” ist somit die Verlegenheitsbezeichnung des Umstandes, dass man trotz Überprüfung aller denkbaren Todesursachen die tatsächliche Ursache für das Versterben des Kindes nicht herausfinden konnte.
In den Jahren nach der Entdeckung des MCAD-Mangels als eigenständige Stoffwechselstörung, aber vor der Einführung des erweiterten Neugeborenenscreenings, war den Eltern der bei ihrem Kind vorliegende MCAD-Mangel gänzlich unbekannt, sodass sie eine während der Nacht eintretende Entgleisung (aufgrund einer aus heutiger Sicht viel zu großen Nahrungspause – oftmals noch dazu in Verbindung mit einer Krankheitssituation wie Erbrechen oder Durchfall) völlig unvorbereitet traf. In einigen Fällen war eine Entgleisung so früh und so heftig eingetreten, dass das Kind bis zum Auffinden durch die Eltern bereits verstorben war, was von den Ärzten dann zunächst meist als plötzlicher, unerwarteter Kindstod angesehen wurde. In der folgenden Autopsie konnte dann aber anhand des Acylcarnitinprofils, bzw. der Genanalyse, nachträglich die Diagnose MCAD-Mangel gestellt und als Todesursache angesehen werden. Im Rückblick auf die Zeiten vor der Entdeckung des MCAD-Mangel kam man damit zu dem Schluss, dass auch damals schon ein gewisser Anteil der zu jener Zeit noch als unerklärlicher plötzlicher Kindstod diagnostizierten Todesfälle ebenfalls von einem unbekannten MCAD-Mangel verursacht worden sein dürften.
Insofern stimmt es schon, dass der MCAD-Mangel etwas mit dem plötzlichen Kindstod zu tun hat(te), allerdings wurde genau dieses Risiko durch die frühzeitige Diagnose dieser Stoffwechselstörung und die sich daraus ergebende erhöhte Wachsamkeit der Eltern weitestgehend beseitigt.
Leider machen manche (kurze) MCAD-Mangel-Definitionen auf den Webseiten einiger Uni-Kliniken keine Unterscheidung zwischen den vollkommen unterschiedlichen Situationen vor und nach der Einführung des erweiterten Screenings, denn da heißt es lapidar, dass zu den Symptomen des MCAD-Mangels neben Krampfanfällen, Unterzuckerungen, Hypoketonurie, Koma, usw. auch der plötzliche Kindstod gehören kann. Somit entsteht der falsche Eindruck, dass das eigene Kind vielleicht sogar trotz Einhaltung kurzer Fütterungsabstände sowie aller anderen Sicherheitsmaßnahmen, einfach aufgrund der Tatsache des bestehenden MCAD-Mangels, ganz plötzlich und unerwartet von einer Minute zur anderen während des Schlafs versterben könnte.
Dieser falsche Eindruck wird verstärkt durch vereinzelt zu findende Berichte, wie z.B. eine Dissertation zum SIDS im Bereich der Rechtsmedizin, die ohne den MCAD-Mangel auch nur ein einziges weiteres Mal im gesamten Text zu behandeln, die Aussage stehen lässt, es gäbe Genmutationen, die den plötzlichen Kindstod direkt verursachten, wie z.B. Defekte des Medium-Chain AcylCoA Dehydrogenase-Gens. Wumms! Hat der MCAD-Gendefekt also doch ein regelrechtes Todesgen produziert, welches völlig ohne Vorwarnung und von einer Minute zur anderen das Leben eines Säuglings ausknipsen kann?
Nein! Erst im Zusammenhang der weiteren Ausführungen wird klar, dass damit dann doch nur gemeint war, dass es sich beim MCAD-Gendefekt um einen aus einer ganzen Reihe möglicher Gendefekte mit Auswirkungen auf den Stoffwechsel handelt, dessen spezielle Wirkung im Bereich der Energiebereitstellung unter gewissen Umständen zu einem lebensbedrohlichen Energiemangel führen kann. Also genau das, was allgemein bekannt ist, und was es mit etwas erhöhter Aufmerksamkeit zu verhindern gilt. Diesen Risikofaktor eines unter gewissen Umständen auftretenden Energiemangelzustands weisen Kinder ohne MCAD-Mangel, bzw. ohne ähnliche den Stoffwechsel beeinträchtigende Gendefekte natürlich nicht auf. Unter diesem − und nur unter diesem − Gesichtspunkt ist die in der obigen Frage enthaltene Behauptung zutreffend.
Eine weitere Dissertation, die so ganz am Rande auch den Einfluss des MCAD-Mangels auf den plötzlichen Kindstod zu beleuchten versucht, kommt zu dem Ergebnis, dass der MCAD-Mangel keine besondere Rolle spielen dürfte − allerdings hat man zum Ableiten dieser Aussage auch nur nach drei (im Verhältnis zur sehr häufigen K329E sehr seltenen) anderen Mutationen bei den verstorbenen Kindern gesucht. Keine davon wurde gefunden, was eigentlich naheliegend war.
Was soll man nun also davon halten, wenn auf den Webseiten mancher Uni-Kliniken so beunruhigende Aussagen stehen, dass der MCAD-Mangel u.a. auch zum plötzlichen Kindstod führen könne und den Eltern das dann auch wieder und wieder von dem sie beratenden Stoffwechselexperten in Erinnerung gerufen wird? Alle diese Aussagen beruhen weitestgehend auf reinen Vermutungen und nicht auf genau belegten Forschungsergebnissen. Weder die oben erwähnte erste, noch die zweite Dissertation gehen bzgl. des Einflusses des MCAD-Mangels auch nur ansatzweise in die Tiefe und liefern verlässliche Ergebnisse. Das einzige, was man mit Sicherheit sagen kann, ist, dass es in der Zeit vor dem Screening höchstwahrscheinlich auch einige an einer aufgrund des MCAD-Mangels im Schlaf eingetretenen metabolischen Krise verstorbenen Kleinkinder gab, deren Todesursache nicht gefunden wurde, da man den MCAD-Mangel zu dem Zeitpunkt noch nicht kannte und daher auch nicht bei der Ursachensuche in Betracht zog.
Es besteht aber kein Grund zur Annahme, dass der MCAD-Mangel das plötzliche Versterben eines ansonsten gerade rundum gesunden Kleinkindes verursachen könnte. Somit gibt es auch keinen Anlass zur übermäßigen Besorgnis! Nicht alles, was man dir diesbezüglich in der Stoffwechselambulanz als scheinbare Fakten mitteilt, ist tatsächlich noch aktuell, geschweige denn wissenschaftlich genau belegt.
Wenn du dies hier liest, bedeutet dies außerdem, dass du um den MCAD-Mangel deines Kindes weißt, und alleine dadurch ist dieser früher nur aufgrund seines Unbekanntseins bestehende Risikofaktor für einen plötzlichen Kindstod schon so gut wie ausgeschaltet. Bei Einhaltung der weiteren Vorsichtsmaßnahmen, die zur Vermeidung eines SIDS empfohlen werden (möglichst nicht Bauch- sondern Rückenlage, Zimmer und Schlafkleidung nicht zu warm, um einer Überhitzung vorzubeugen, keine weichen Kissen oder Decken, die das Gesicht des Kindes bedecken können, usw.) ist das Risiko generell sehr gering, und jedenfalls nicht höher, als bei jedem anderen Kind auch. Verschiedene Untersuchungen haben festgestellt, dass generell das anscheinend geringste Risiko bei denjenigen Kindern besteht, die in einem eigenen Bett im Elternzimmer schlafen. Auch aufgrund der Notwendigkeit des regelmäßigen nächtlichen Fütterns, bietet sich daher z.B. ein am Elternbett befestigter Babybalkon als gut geeignete Lösung an.![]()
Nein, dein Kind wird nicht automatisch dick werden − zumindest nicht wegen des MCAD-Mangels! Auch wenn Dir das vielleicht von deinem Stoffwechselarzt oder Ernährungsberater an der Uni-Klinik schon kurz nach der Geburt deines Kindes so in Aussicht gestellt wurde, ist diese Behauptung keineswegs zutreffend.
Der Vorgang des bei Bedarf erfolgenden Herauslösens der Fettsäuren aus dem Fettgewebe des Körpers funktioniert bei deinem Kind wie bei jedem anderen auch. Das hat noch überhaupt nichts mit dem MCAD-Mangel zu tun. Die freigesetzten Fettsäuren werden durch das Blut zur Leber transportiert. Innerhalb der Leberzellen gelangen sie dann in die Mitochondrien – und genau hier kommt es erst zu der Auswirkung des MCAD-Mangels, nämlich zu der unvollständigen Zerlegung der Fettsäuren in kleinere Bausteine, die dann als Energieträger für andere Organe dienen können.
Die Auswirkung des MCAD-Mangels besteht somit nicht darin, dass dein Kind seine im Fettgewebe gespeicherten Fettreserven nicht verwerten könnte, sondern vielmehr darin, dass bei der Verwertung dieser Fettreserven innerhalb der Mitochondrien nur ein geringer Anteil der gespeicherten Energie wieder freigesetzt wird, um deinem Kind zur Deckung seines Energiebedarfs zur Verfügung zu stehen. Während jedes Gramm reines Fett normalerweise einen Energiegehalt von etwa 9,3kcal enthält, die beim Abbau für die Versorgung des Körpers (vor allem des Gehirns) wieder freigesetzt werden, kann ein Kind mit MCAD-Mangel davon in gesunden Zeiten nur etwa die Hälfte nutzen. Im Fettgewebe gespeichertes Fett enthält übrigens pro Gramm Masse “nur” etwa 7kcal, da bei der Einlagerung in die Fettzellen auch gleichzeitig Wasser mit gebunden wird, das somit einen Teil der Gesamtmasse ausmacht, aber keinen Energieträger darstellt.
Das bedeutet aber nicht, dass es ihm daher besonders schwer fiele, sein Gewicht zu halten, oder gar zu reduzieren. Möglicherweise trifft sogar das Gegenteil zu! In dem Artikel “11. Fragen an die Stoffwechselexperten” habe ich eine Theorie formuliert, nach der es Kindern mit MCAD-Mangel sogar wesentlich leichter fallen dürfte, normalgewichtig zu bleiben, als allen anderen Kindern, da zwar auch bei ihnen der Fettanteil aus der Currywurst mit Pommes zunächst mal in den Fettreserven landet, dieses “Pölsterchen” aber mit der Hälfte des Aufwands, den Kinder ohne MCAD-Mangel treiben müssten, wieder abgebaut werden kann, da die verbleibende Hälfte jeder einzelnen Fettsäurenkette nicht weiter genutzt werden kann, sondern ausgeschieden werden muss.
Selbst wenn diese Theorie nicht zutreffen sollte (dazu wurden anscheinend bislang noch keine gezielten Untersuchungen angestellt), zeigt das Ergebnis der bayerischen Langzeitbeobachtung der an der betreffenden Studie teilnehmenden Kinder und Jugendlichen mit MCAD-Mangel, dass der unter ihnen zu findende Anteil normalgewichtiger Kinder unter 10 Jahren vollkommen mit dem deutschen Durchschnitt (ca 85%) übereinstimmt, und es auch nicht mehr leicht und stark übergewichtige Kinder gibt, wie unter ihren Altersgenossen. In der Altersgruppe der Jugendlichen war der Anteil der normalgewichtigen Kinder mit MCAD mit 91,5% sogar deutlich über dem deutschen Durchschnitt von 85,3%. Diese Zahlen sind inzwischen schon einige Jahre alt. Sie wurden von den Verantwortlichen der Langzeitbeobachtung auf einer in München durchgeführten Informationsveranstaltung für vom MCAD-Mangel betroffene Familien als bereits eine der wesentlichen und sehr positiven Erkenntnisse der Studie genannt.
Falls du dich an der Studie beteiligen möchtest (indem du von Zeit zu Zeit Fragebögen zur Entwicklung deines Kindes ausfüllst), kannst du dich an die auf der Informationsseite des LGL zum Neugeborenenscreening genannte Mitarbeiterin zur Langzeitstudie wenden – und ihr gerne einen herzlichen Gruß von mir ausrichten :). Obwohl es sich um eine Studie des Bayerischen Landesamtes für Gesundheit und Lebensmittelsicherheit (LGL) handelt, können sich trotzdem auch MCAD-Betroffene aller anderen Bundesländer beteiligen.
Vielleicht kann man das als Indiz für die Richtigkeit obiger Theorie ansehen, vielleicht liegt es auch einfach nur daran, dass die Eltern der vom MCAD-Mangel betroffenen Kinder von Anfang an auf eine gesunde und ausgewogene Ernährung der Kinder zu achten versuchen. Wie weit sich das tatsächlich realisieren lässt, ist eine ganz andere Frage. Auf jeden Fall zeigt dieses Ergebnis deutlich, dass der MCAD-Mangel keinesfalls dazu führt, dass davon betroffene Kinder im Lauf ihres Lebens zwangsläufig immer dicker und dicker werden, sondern dass sie sich völlig normal entwickeln. Diese Feststellung wurde auch durch die körperliche Entwicklung der über viele Jahre hinweg im früheren MCAD-Forum vertretenen Kinder bestätigt. Mit wenigen Ausnahmen haben sie sich alle zu völlig normalgewichtigen und dabei teilweise sehr sportlichen Teenies, Jugendlichen und Erwachsenen hin entwickelt.
Was für “wenige Ausnahmen”? Wie jedes andere Kind, kann selbstverständlich auch ein Kind mit MCAD-Mangel etwas pummelig oder sogar sehr dick werden, wenn es übermäßig viel Nahrung zu sich nimmt, weil es von seinen besorgten Eltern weit über Bedarf mit Naschereien und kohlenhydrathaltigen oder fettreichen Lebensmitteln versorgt wird. Ein Kind mit MCAD-Mangel benötigt aber nicht MEHR Nahrung als andere Kinder, denn es verbraucht aufgrund seiner Aktivitäten auch nur genau so viel Energie wie jedes andere Kind auch. Wichtig ist deshalb nicht eine größere Nahrungsmenge, sondern dass die gleiche Tages-Energiemenge einfach nur in regelmäßigen Intervallen zugeführt wird und keine zu großen Pausen dazwischen liegen.
Der Fettanteil der täglichen Speisen sollte gegenüber dem, was man als Familie sonst vielleicht als normal betrachtet, ein wenig zugunsten des Kohlenhydratanteils reduziert sein. Wenn man den Fettanteil nämlich unverändert beibehält, aber den Kohlenhydratanteil noch zusätzlich durch Malto, Speisestärke oder zuckerhaltige Limos aufstockt, wird dem Kind insgesamt zu viel Energie zugeführt und dann können sich auf Dauer auch deutlich sichtbare Speckpolster aufbauen. Dies liegt dann aber, wie oben beschrieben, nicht am MCAD-Mangel, sondern an einer fortwährenden Überfütterung.
Eines darf man nämlich bei dieser ganzen komplizierten Fettthematik nicht verwechseln: Dein Kind hat zwar ein Problem mit der Fettverwertung, also der Wiedergewinnung der darin enthaltenen Energie − es hat aber nicht das geringste Problem bei der Fettaufnahme und -einlagerung. Und wie viele Menschen selbst aus leidvoller Erfahrung wissen: Fett kommt üblicherweise, um zu bleiben.![]()
Nein, den MCAD-Mangel selbst kann dein Kind überhaupt nicht weitervererben! Es selbst hat den MCAD-Mangel auch nur dadurch bekommen, dass es von seinen beiden Eltern jeweils deren eine defekte Genkopie vererbt bekam. Da jeder Mensch von jeder genetischen Anlage immer zwei Kopien besitzt (eine auf dem von der Mutter geerbten Chromosomenanteil und eine auf dem väterlichen Anteil) enthalten die eigenen Spermien oder Eizellen dieses Menschen immer eine Mischung sämtlicher Anlagen in einfacher Ausprägung, also entweder die eine Genkopie oder die andere. In diesem Zusammenhang verwendet man auch den Begriff “Allel”. Das eine Allel für ein bestimmtes körperliches Merkmal kommt ursprünglich von der Mutter, das andere vom Vater.
Dein Kind wird daher immer die Anlage zum MCAD-Mangel vererben, denn da bei ihm beide Allele einen MCAD-Gendefekt aufweisen, werden deshalb auch alle seine Spermien oder Eizellen entweder das eine oder das andere von dem Gendefekt betroffene Allel transportieren. Kinder mit der Mutationskombination K329E homozygot können an ihre eigenen Kinder immer nur genau diese Anlage vererben − was anderes haben sie ja nicht, da beide Allele den gleichen Gendefekt aufweisen, sodass ihre Kinder wenigstens Träger (Carrier) dafür werden. Kinder mit compound heterozygoten Mutationskombinationen (z.B. K329E/Y67H) werden entweder das Allel mit dem schweren Defekt (K329E) oder dem leichten Defekt (Y67H) vererben, und es hängt einzig und alleine davon ab, welche dieser beiden Allel-Varianten in dem Sieger-Spermium oder in der gerade reifen Eizelle enthalten sein wird.
Für die andere Genhälfte deiner Enkel ist der spätere Partner deines Kindes verantwortlich. Und da stehen die Chancen sehr gut, dass dieser ihm keine zweite Anlage vererbt. Schließlich ist nur rund jede 60. Person in Deutschland Carrier eines MCAD-Gendefekts. Somit können 59 von 60 Frauen oder Männer gar keine MCAD-Mangel-Anlage vererben, d.h. von einer solchen Mutter oder einem solchen Vater werden deine Enkel nur ein intaktes Gen vererbt bekommen und damit selbst nur Träger sein, so wie fast alle MCAD-Eltern weltweit. “Fast alle” deshalb, weil es einen sehr kleinen Anteil an Elternteilen gibt, die selbst einen MCAD-Mangel haben, weil zufälligerweise auch ihre beiden Eltern schon Carrier waren und ihnen beide jeweils diese defekte Genkopie vererbt hatten.
Sollte dein Kind aber ausgerechnet mit diesem 60. Mann oder dieser 60. Frau, die selbst Carrier sind, ein Kind zeugen, dann besteht eine Chance von 50%, dass das Kind von diesem Elternteil das für die Ausbildung des MCAD-Mangels benötigte zweite defekte Gen vererbt bekommt.
Die Gesamtwahrscheinlichkeit für diesen Fall ist somit 1/60 • 1/2 = 1/120, und das sind 0,83%. Umgekehrt wird es deutlicher: mit 99,17%iger Wahrscheinlichkeit werden die Kinder deines Kindes keinen MCAD-Mangel haben.
In der Generation deiner Urenkel kann bei einem Teil der Nachkommen sogar die Anlage für den MCAD-Mangel wieder vollständig aus dem Erbgut verschwunden sein, wenn nämlich deine Enkel an ihre Kinder den intakten Chromosomenanteil vererben, und diese vom anderen Elternteil auch ein Chromosom mit intaktem MCAD-Gen erhalten.
Ein paar Eltern haben in der Vergangenheit zwar schon von ihrem Arzt gesagt bekommen, dass ihr Kind seinen MCAD-Mangel höchstwahrscheinlich auch an seine eigenen Kinder weitergeben würde, jedoch ist diese Aussage, wie oben gezeigt, nicht zutreffend. Es gibt lediglich die Anlage weiter, jedoch nicht die vollständige Stoffwechselstörung. Anderen Eltern wurde von ihren Ärzten gesagt, dass ihr Kind die gleiche Stoffwechselstörung habe wie sie selbst − die Eltern aber nur in einer leichten Form und das Kind in einer schweren. Auch das ist, wie oben gezeigt, nicht korrekt und zeigt nur, dass den betreffenden Ärzten das beim MCAD-Mangel zwingend zu berücksichtigende Prinzip der rezessiven Vererbung selbst nicht klar ist. Möglicherweise hängt diese falsche Ansicht auch mit dem vermischten und somit missverständlichen Gebrauch des Begriffs “MCAD-Defekt” zusammen, den manche Ärzte leider anstelle der korrekten Bezeichnung MCAD-Mangel benutzen − wodurch andere, mit diesem Gebiet selbst wenig vertraute Ärzte verwirrt werden. Ja, einer der beiden MCAD-(Gen-)Defekte eines Menschen mit MCAD-Mangel wird in jedem Fall an die eigenen Kinder vererbt, sodass diese zwingend auch einen “MCAD-Defekt” (also die Anlage!) haben! Allerdings ist das dann nur ein einseitiger Gendefekt und noch lange kein MCAD-Mangel, denn der erfordert zwingend einen zweiten geerbten “MCAD-Defekt” vom anderen Elternteil. Ein Grund mehr, weshalb sowohl Ärzte als auch Eltern von Anfang an darauf achten sollten, diese Begriffe sauber auseinanderzuhalten. Auf dieser und allen folgenden Seiten wird daher niemals vom MCAD-Defekt, sondern ausschließlich vom MCAD-Mangel gesprochen, um diesen Unterschied ganz klar herauszustellen und dich an die Vermeidung des Begriffs MCAD-Defekt zu gewöhnen.![]()
Vor einigen Jahren formulierte eine Teilnehmerin ihre beim Lesen anderer Forumsbeiträge aufgekommenen Sorgen mit folgendem Satz:
| “Wenn ich sonst so im Forum stöbere, lese ich die vielen Berichte über Stoffwechsel-Entgleisungen und mir ist es bisher so vorgekommen, als ob alle Kinder mit MCAD-Mangel regelmäßig entgleisen.” |
Damit war sie bei weitem nicht die einzige. Auch andere Teilnehmer − vor allem erst neu mit der MCAD-Diagnose konfrontierte Eltern − hatten sich teilweise sehr schnell wieder aus dem Forum zurückgezogen, weil sie sich durch die (vermeintlich) häufig von Entgleisungen handelnden Beiträge der anderen Teilnehmer stark beunruhigt fühlten. Einige schrieben mir zum Abschied sinngemäß, sie könnten es psychisch nicht ertragen, davon zu lesen, dass anscheinend viele andere Kinder immer wieder entgleisten, weil es sie zu traurig und ängstlich bzgl. der Zukunftsaussichten ihres eigenen Kindes mache.
Dieser Eindruck täuschte jedoch. Von den im Verlauf der gesamten zehnjährigen Forumszeit insgesamt rund 550 registrierten MCAD-Teilnehmern hatten etwa 530 nie von irgendwelchen riskanten Situationen, geschweige denn Entgleisungen berichtet, auch wenn wirklich hin und wieder von Krankenhausaufenthalten zu lesen war. Das waren aber alles lediglich Vorsichtsmaßnahmen während Krankheitsphasen mit problematischer Nahrungsaufnahme, um von vorneherein auf der sicheren Seite zu sein. Vorsorgliche Krankenhausaufenthalte mit Infusionen gehören beim MCAD-Mangel zur Prävention nun mal leider dazu. Es bedeutete aber fast nie, dass es schon zu Ansätzen einer Entgleisung gekommen war, sondern als Eltern fühlt man sich damit − zumindest in den ersten paar Jahren − einfach sicherer.
Dass trotzdem der Eindruck entstand, hier wäre sehr oft und von vielen Entgleisungen geschrieben worden, war vielmehr ein Beispiel selektiver Wahrnehmung. Durchstöberte man das Forum mittels der Suchfunktion nach dem Begriff “Entgleisung”, wurde man auch fündig, denn es gab natürlich schon ein paar Mitglieder, die davon berichtet hatten. Dazu gehörten 4-5 Familien mit bereits älteren Kindern (oder selbst vom MCAD-Mangel betroffene Mitglieder), auf deren MCAD-Mangel man vor längerer Zeit durch das Eintreten einer Entgleisung überhaupt erst aufmerksam wurde. Hätte man es bereits von Anfang an gewusst, wäre es durch die dann gesteigerte Alarmbereitschaft und Vorsicht möglicherweise niemals dazu gekommen. Aus diesem Grund waren diese berichteten Fälle mit der heutigen Situation nicht vergleichbar. Es waren aus den Reihen und dem Bekanntenkreis der Mitglieder auch drei am MCAD-Mangel verstorbene Kinder bekannt, aber auch bei diesen war der MCAD-Mangel leider nicht rechtzeitig diagnostiziert worden.
Darüber hinaus waren hier aber tatsächlich auch ein paar Kinder vertreten, deren MCAD-Mangel im NG-Screening frühzeitig erkannt wurde, und die trotzdem schon ein oder zwei Situationen erlebt hatten, in denen eine beginnende Entgleisung festzustellen war. In allen diesen Fällen konnten die Eltern das bereits etwas apathisch wirkende Kind aber mit Zuführung von ein wenig Saft, Maltolösung oder Traubenzucker schnell wieder zu einem Normalzustand zurückführen. In dem einen oder anderen Fall schloss sich dann auch ein kurzer Krankenhausaufenthalt an, um das Ergehen des Kindes weiter zu beobachten. Hinter diesen Berichten steckten allerdings nicht viele, sondern nur (man möge dieses “nur” bitte entschuldigen, aber bei manchen Lesern hatte sich ein ziemlich verzerrtes Bild bezüglich der Mengenrelationen gebildet) 5-6 Mitglieder, die aber in einer ganzen Reihe von Forumsbeiträgen mehrfach von ihren diesbezüglichen Erlebnissen berichtet hatten. Da man als neues Mitglied meist noch nicht den Überblick hatte, zu wem welches Kind gehörte, erkannte man auch nicht ohne weiteres, dass es sich dabei immer um die gleichen 5-6 Entgleisungssituationen handelte, und auch nur Kinder mit der bekannten Risikovariante K329E homozygot, oder (in einem Fall) einer vergleichbar schweren Mutationskombination betroffen waren. Wie gesagt soll das Erlebnis der betroffenen Mitglieder mit dem aufgestellten Mengenvergleich (6 von 550 sind 1,1%) nicht verharmlost, geschweige denn als “die paar Fälle zählen doch nicht” abgetan werden. Es soll nur verdeutlichen, dass sich durch das frühzeitige Feststellen des MCAD-Mangels sehr vieles zum Besseren gewendet hat und es selbst bei Vorliegen der Risikovariante nur noch zu extrem wenigen Fällen echter Entgleisungen kommt − und selbst diese können aufgrund der allzeit gesteigerten Aufmerksamkeit der informierten Eltern in den meisten Fällen schnell selbst behandelt und schon im Anfangsstadium unterbrochen werden.
Im Übrigen waren es oft gar nicht die Berichte der anderen betroffenen Familien, sondern die Schilderungen der betreuenden Stoffwechselärzte, die bei den Eltern die heutzutage längst nicht mehr so begründete Angst vor der Stoffwechselentgleisung oder gar -krise schürten. Immer wieder berichteten Teilnehmer davon, dass sie bei jedem der regelmäßigen Kontrolltermine neue Geschichten von Entgleisungsfällen oder gar durch den MCAD-Mangel verursachten Todesfällen aufgetischt bekamen, sobald sie gegenüber ihrem Arzt auch nur ansatzweise zu verstehen gaben, dass sie sich inzwischen im Umgang mit dem MCAD-Mangel ihres Kindes schon relativ sicher fühlten. Leider blieb bei diesen erschreckenden Schilderungen ihnen gegenüber meistens der wichtige Punkt unerwähnt, ob es sich dabei um Entgleisungen von bereits frühzeitig diagnostizierten Kindern handelte, die trotz der erhöhten Aufmerksamkeit ihrer Eltern eingetreten waren (heutzutage sehr selten), oder ob es sich dabei immer noch um Personen handelte, deren MCAD-Mangel all die Jahre bis zum Eintritt dieser auslösenden Situation völlig unbekannt war. Dies dürften auch heute noch die meisten ernsten Entgleisungsfälle sein, aber diese Fälle sind, wie oben schon erwähnt, nicht mit der Situation der seit einigen Jahren frühzeitig diagnostizierten Kinder und Jugendlichen vergleichbar und sollten daher nicht ständig neu zur anscheinend für notwendig erachteten “Verunsicherung” der sich langsam sicher fühlenden Eltern herangezogen werden.![]()
Es kann natürlich niemals hundertprozentig ausgeschlossen werden, dass irgendwelche “Auffälligkeiten” doch irgendwie mit dem MCAD-Mangel zu tun haben könnten, aber das ist sehr unwahrscheinlich. Es kommt allerdings nicht selten vor, dass man im Austausch mit anderen MCAD-Betroffenen auf von mehreren Beteiligten gemachte Beobachtungen stößt, die einen dann schnell zu dem Schluss verleiten, dies habe mit dem sie alle verbindenden Thema − in diesem Fall dem MCAD-Mangel − zu tun. Wissenschaftlich nennt man dieses Phänomen den sogenannten “ascertainment bias”, was auf Deutsch mit Erhebungsbefangenheit oder -verzerrung übersetzt werden könnte. Damit ist zunächst mal gemeint, dass die Untersuchungsergebnisse bzgl. der Häufigkeit von irgendwelchen Vorkommnissen davon beeinflusst werden können, in welchem “Erhebungsumfeld” man die Untersuchung durchführt. Dadurch kann das Ergebnis einer Studie drastisch verzerrt werden.
Ganz oben auf dieser Seite hatte ich z.B. bereits die jährlich vielen falsch-positiven Ergebnisse im Neugeborenen-Screening in Bezug auf MCAD erwähnt. Uns gegenüber behauptete damals der Stoffwechselarzt, das NG-Screening liefere absolut verlässliche, weil zu fast 100% korrekte Ergebnisse. Der Anteil der falsch-positiven Befunde sei lediglich im Promille-Bereich. Daher gäbe es alleine schon aufgrund der Auffälligkeit im NG-Screening keinerlei Zweifel mehr am vorliegenden MCAD-Mangel. Sein Fehler an der Stelle war, außer Acht zu lassen, dass sich diese von ihm irgendwo gelesene fast 100%ige Korrektheit auf die Anzahl der falsch-positiven Ergebnisse über alle Zielkrankheiten hinweg und auf alle pro Jahr gescreenten Kinder bezog. Unter den damals (2007) rund 700.000 jährlich gescreenten Kindern waren insgesamt 700 Kinder mit falsch-positiven Befunden tatsächlich nur 0,1%. Der Stoffwechselarzt hätte in unserem konkreten Fall bei seiner Aussage aber bedenken müssen, dass dies die falsche Bezugsgruppe war, weil es unter den wenigen überhaupt in Richtung MCAD-Mangel im Screening auffällig gewordenen Kindern damals (2007) generell noch rund 62% falsch-positive Befunde gab. Diese Untergruppe von jährlich rund 170 Kinder wäre das korrekte Erhebungsumfeld gewesen und nicht die Gesamtanzahl aller pro Jahr geborenen Kinder, von denen rund 99% ohnehin nicht im Screening auffallen. Ein typischer Fall von angewandtem ascertainment bias.
Ein anderes Beispiel erlebte ich vor einigen Jahren, als ich mich vorübergehend auch mal in der Facebook-Gruppe der amerikanischen Selbsthilfegruppe (fodsupport.org) beteiligte, in der Hoffnung vielleicht auch von dort aus neue Informationen in das hiesige Forum einbringen zu können. Damals berichtete dort eine Mutter davon, dass ihr Kind beim Baden selbst im warmen Wasser immer irgendwann blaue Lippen bekäme und am ganzen Körper zittere. Es fanden sich dann innerhalb von wenigen Stunden noch etwa zehn weitere Teilnehmerinnen (von damals über 1000 aktiven Mitgliedern der amerikanischen Facebook-Gruppe), die bei ihrem Kind das gleiche Phänomen festgestellt hatten und irgendjemand brachte dann die Vermutung, dass das für ihn irgendwie nach einem Energiemangel klinge, ins Spiel. Daraufhin kam jemand zu der Schlussfolgerung: “Unfassbar. Ich hätte nie gedacht, dass das mit dem MCAD-Mangel zu tun hat. Aber jetzt leuchtet mir das alles ein!”. Diese Feststellung wurde in kürzester Zeit von allen sich an der Diskussion beteiligenden Personen als nun endlich erwiesene Tatsache bestätigt. Ich gab diese Story hier im Forum weiter und auch hier konnten zwei oder drei der Teilnehmer von identischen Beobachtungen berichten. Was lag also näher, als es tatsächlich für wahrscheinlich zu halten, dass die blauen Lippen und das Zittern eine Folge des MCAD-Mangels wären. Ich stellte diese Frage aber auch noch in einem allgemeinen Elternforum mit mehreren tausend TeilnehmerInnen. Auch dort konnten direkt fast 20 Eltern davon berichten, dass ihre Kinder haargenau die gleichen Symptome beim Baden zeigten und sie hätten sich auch schon lange gefragt, woher das nur käme − aber keines dieser Kinder hatte einen MCAD-Mangel! Damit war klar, dass es nichts mit dieser Stoffwechselstörung zu tun hatte, sondern einfach querbeet anzutreffen war. Diese Neuigkeit wurde hier im Forum auch noch offen aufgenommen und akzeptiert − so richtig daran geglaubt hatte ohnehin niemand. Als ich aber danach auch in der amerikanischen Facebook-Selbsthilfegruppe dieses Ergebnis mitteilte, wurde ich regelrecht in der Luft zerrissen. Die beteiligten Eltern waren so froh gewesen, eine dermaßen einfache Erklärung für diese sie verwundernde Beobachtung gefunden zu haben − man wollte es sich nicht nehmen lassen. Sie wollten unter allen Umständen daran glauben, dass es einzig und alleine mit dem MCAD-Mangel zu tun habe. Selbst andere Teilnehmer, die sich zuvor in diesem Thema nicht zu Wort gemeldet hatten, bestätigten die aufgebrachten Eltern darin, dass mein dagegen sprechender Einwand völliger Blödsinn sei, und sie selbst jetzt ebenfalls absolut sicher wären, dass es einzig und alleine eine Auswirkung des MCAD-Mangels sei. In diesem Fall bestand der ascertainment bias also darin, dass man sich eine Beobachtung gegenseitig in einer geschlossenen Gruppe bestätigte und dabei zu dem Schluss kam, es sei ein Phänomen, das NUR innerhalb dieses Erhebungsumfeldes auftrete. Dass es außerhalb der Gruppe mit der gleichen Häufigkeit vorkommt und daher nichts mit dem die Gruppe verbindenden Element − dem MCAD-Mangel − zu tun hatte, wurde zuerst übersehen und schließlich bewusst ausgeblendet, weil den beteiligten Personen das von ihnen selbst “gefälschte” Testergebnis so gut in den Kram passte.
Durch speziell dieses zweite Beispiel soll verdeutlicht werden, dass es auch beim Austausch über den MCAD-Mangel dazu kommen kann, dass man ihn als Ursache für Auffälligkeiten und Beobachtungen heranzieht, mit denen er überhaupt nichts zu tun hat. Im gegenseitigen Austausch ist somit grundsätzlich ein wenig Vorsicht und Nüchternheit angeraten.![]()
Leider nein, so schön und angenehm es auch wäre. Genetisch bedingte Stoffwechselstörungen gehören zu einer Klasse von Krankheiten, gegen die man größtenteils nicht medikamentös vorgehen kann. Es gibt zwar aktuelle Bestrebungen in den Forschungsbereichen der Bio- und Gentechnik, die Funktionsfähigkeit der Enzyme bei z.B. PKU und MCAD-Mangel durch hoch dosierte Zuführung sogenannter “Kofaktoren” zu verbessern, jedoch betrifft dies nur solche Ausprägungen, bei denen die Einschränkungen der Enzymfunktion auf eine falsche Proteinfaltung zurückzuführen sind. Darüber hinaus wurden erste Erfolge bislang fast nur bei den “milden” Ausprägungen der PKU festgestellt, während die schweren Ausprägungen nur in Einzelfällen und dann auch nur geringfügig auf die Behandlung ansprachen.
Ich hatte auch schon einmal vorgehabt, an dieser Stelle auf die Möglichkeiten einzugehen, die sich durch das 2020 mit dem Chemie-Nobelpreis ausgezeichnete “Genschere”-Verfahren “CRISPR/Cas9” eröffnen, denn dadurch könnten sich in Zukunft medizinische Einsatzmöglichkeiten der Genbearbeitung und -korrektur jenseits allen bisherigen Denkens auftun. Aber auch wenn damit − theoretisch − beliebige genetisch bedingte Erbkrankheiten wie ja auch der MCAD-Mangel im menschlichen Körper vollständig behoben, um nicht zu sagen ausradiert werden könnten, ist die Forschung noch lange nicht so weit, dieses Verfahren in Form eines zuverlässig wirkenden Medikaments auf den Markt zu bringen. Man will schließlich nicht riskieren, durch eine bewusst herbeigeführte Genmanipulation im Erbgut, einen MCAD-Mangel (vielleicht) zu beseitigen und versehentlich ein eigentlich stillgelegtes Krebsgen zu aktivieren. Und selbst wenn es in ein paar Jahren wirklich möglich sein sollte, eine genetische Mutation innerhalb der DNA so sicher wie in einem Textverarbeitungsprogramm einfach durch Löschen und Ersetzen mit den richtigen Buchstaben zu korrigieren, stehen der praktischen medizinischen Anwendung immer noch jede Menge Hürden in Form geltender Gesetze gegen die Genmanipulation entgegen. Daher möchte ich CRISPR/Cas9 an dieser Stelle zwar nicht unerwähnt lassen, aber dabei soll es auch vorerst bleiben. Wen das Thema aber weiter interessiert, kann z.B. auf den Seiten der Max-Planck-Gesellschaft nähere Informationen dazu finden.
Zurück zu den gezielten wissenschaftlichen Bestrebungen zur Behandlung des MCAD-Mangels. Bereits vor einigen Jahren wurde in ein paar Studien eine leichte positive Wirkung einer auf lange Zeit angelegten täglichen Gabe von Riboflavin (Vitamin B2) auf die Aktivität des MCAD-Enzyms beobachtet, allerdings sind diese gemessenen Aktivitätssteigerungen bislang vor allem für die Forscher interessant, haben aber noch keine nennenswerte Bedeutung für das tägliche Leben der selbst vom MCAD-Mangel betroffenen Menschen. Riboflavin ist somit nicht als Medikament zu betrachten, denn es hilft keineswegs, einen schweren MCAD-Mangel in einen milden zu verwandeln, oder gar vollständig zu heilen.
Es bleibt darüber hinaus übrigens zweifelhaft, ob es jemals ernsthafte Bestrebungen geben wird, ein auf welche Weise auch immer den MCAD-Mangel heilendes oder zumindest unterdrückendes Medikament zu entwickeln und in den Handel zu bringen. Dazu gibt es einfach zu wenige Betroffene in der Bevölkerung, und deren Behandlung ist zu kostengünstig, wenn man es mal mit den Betroffenen anderer Stoffwechselstörungen vergleicht, für deren Überleben fortwährend sehr aufwändige Behandlungen notwendig sind, die die Krankenkassen sehr viel Geld kosten. Menschen mit MCAD-Mangel müssen für ein weitgehend unbeschwertes Leben dagegen “nur” auf regelmäßige Nahrungsaufnahme achten, und die paar präventiven Glukose-Infusionen während schwieriger Krankheitsphasen rechtfertigen für niemanden die Investition hoher Summen in die Entwicklung eines dann womöglich selbst noch extrem teuren und dauerhaft einzunehmenden Medikaments, das in der Lage wäre, die Auswirkungen des MCAD-Mangels ein wenig abzuschwächen − Auswirkungen, die im normalen, gesunden Alltag ohnehin nicht spürbar sind. Auch das inzwischen nur noch von wenigen Stoffwechselärzten zur täglichen Einnahme verordnete Carnitin fällt mit seinem relativ hohen Preis da nicht wesentlich ins Gewicht.
Man braucht somit vermutlich keine Hoffnungen in ein in näherer Zukunft auf den Markt kommendes Medikament zu setzen, denn auch selbst, wenn die Pharmaindustrie es tatsächlich wollte − auf den Gebieten der Bio- und Gentechnik wird zwar intensiv geforscht, aber bis zur flächendeckenden medizinischen Einsetzbarkeit werden noch viele Jahre vergehen.
Sowohl für Eltern von betroffenen Kindern, als auch für die Betroffenen später selbst, als auch für die mit Stoffwechselstörungen befassten Ärzte ist deshalb das Begreifen der Physiologie, also den mit dem (Fett-)Stoffwechsel in Verbindung stehenden physikalischen und biochemischen Vorgängen in den Zellen und Organen, das A und O, um den MCAD-Mangel erfolgreich behandeln und seine möglichen Auswirkungen vermeiden zu können! Dies ist die wichtigste Feststellung überhaupt, und genau deshalb ist es für jede betroffene Familie von größter Bedeutung, sich selbst ausgiebig über den MCAD-Mangel zu informieren.![]()
Die Frage nach der homöopathischen Behandelbarkeit des MCAD-Mangels wurde mir zugegebenermaßen nicht wirklich häufig, aber solange auf dieser Seite dazu noch nichts stand, doch mit schöner Regelmäßigkeit gestellt. Etwa einmal pro Jahr trat wieder jemand mit dem Thema an mich heran, wollte sich dann aber oftmals mit meiner Rückmeldung nicht zufrieden geben. Ich habe deshalb inzwischen absolut keine Lust mehr darauf zu antworten, nur um dann immer wieder vorgeschwärmt zu bekommen, als was für milde und gleichzeitig wirkungsvolle “Medikamente” man seine ganzen Globuli bisher erlebt habe und dass man den MCAD-Mangel doch auch in diese ganzheitliche Betrachtungsweise einbeziehen müsse. Falls du so ein glühender Verfechter der Homöopathie bist, dann mach doch was du willst, denn dann interessiert dich meine davon abweichende Meinung ohnehin nicht! Wenn du aber im Interesse der Gesundheit deines Kindes ernsthaft wissen möchtest, ob man den MCAD-Mangel damit behandeln kann, dann lies weiter.
Nein! Es gibt keine Möglichkeit, den MCAD-Mangel auch nur ansatzweise homöopahisch zu behandeln! Das wird dir auch jeder Stoffwechselarzt bestätigen. Sollte dir ein Homöopath, Kinesiologe, Kinderarzt oder deine Hebamme, deine Eltern oder jemand aus dem Freundeskreis etwas anderes erzählen und dir irgendwelche Globuli für dein Kind empfehlen, um damit das Risiko von Stoffwechselentgleisungen oder auch nur von Nahrungsverweigerung zu reduzieren, lass dir bitte nichts einreden. Solange dein Kind in normalem Umfang Nahrung zu sich nehmen kann, hat es ohnehin kein großes Risiko, in eine Unterzuckerung und eine sich daraus ergebende Stoffwechselkrise zu geraten. Solche Situationen sind heutzutage, dank der frühen Diagnose des MCAD-Mangels im erweiterten Neugeborenenscreening, etwas sehr seltenes. Es kann natürlich sein, dass du beim Stöbern im Internet oder in der Facebook-Gruppe relativ häufig von Stoffwechselentgleisungen liest, es haben aber nur sehr wenige Betroffene so etwas tatsächlich schon mal erlebt – es wird von ihnen jedoch vergleichsweise oft erwähnt. Demgegenüber steht eine um ein Vielfaches größere Gruppe von Familien, deren Kinder, trotz schwerem MCAD-Mangel, niemals auch nur die geringsten daraus resultierenden Symptome hatten – davon wird nur so gut wie nie berichtet. Wenn du deinem Kind also bereits irgendwelche Globuli gibst und es bisher keine MCAD-Symptome gezeigt hat, dann ist das nicht eine dem homöopathischen Mittel zuzuschreibende Ausnahme, sondern einfach nur der heutzutage zu erwartende Normalfall.
Besonders dann, wenn es zu einer problematischen Krankheitssituation mit Nahrungsverweigerung kommt, darfst du nicht den Fehler machen, deinem Kind erst noch ein paar Globuli dagegen zu geben und abzuwarten, ob es besser wird. Das ist nicht zu erwarten und vergeudet nur wertvolle Zeit. Wenigstens in den ersten Jahren solltest du dich mit deinem Kind schnellstmöglich in eine Kinderklinik oder ins nächste Krankenhaus begeben, um es dort mit Glucoseinfusionen versorgen zu lassen. Wenn du ihm bis dahin noch Globuli geben möchtest, kannst du das natürlich tun – am Besten in großen Mengen und in kurzen Abständen. Immerhin bestehen die Kügelchen ausschließlich aus Zucker und den kann dein Kind dann wirklich brauchen. Dabei wäre es auch vollkommen egal, welche Globuli du ihm dann esslöffelweise in den Mund gibst. Es macht auch überhaupt keinen Unterschied, in welcher Potenz du ihm Cepa, Arnica, Nux Vomica, Belladonna, Aconitum, Gelsemium, Echinacea, oder was die homöopathische Hausapotheke für Kinder sonst noch so zu bieten hat, verabreichst. Damit würden die vielen Gläschen dann auch endlich mal wirklich leer, die man sonst nach einigen Jahren, wenn das “Haltbarkeits”-Datum der Zuckerkügelchen abgelaufen ist, alle in fast ungenutzem Zustand in den Müll, oder besser in den Tee schüttet. Preiswerter wären für eine solche Ausnahmesituation natürlich einfach ein paar Zuckerwürfel, etwas hochdosierte Maltodextrin-Lösung oder andere sehr stark zuckerhaltige Getränke, denn das hätte den gleichen Effekt.![]()
Sicher nimmt dein Kind Carnitin ein, oder? Der zunächst in der Windel, aber möglicherweise auch im Schweiß feststellbare fischige Geruch kommt vom Trimethylamin (TMA). Dabei handelt es sich um die Substanz, die auch alten Fisch zum Müffeln bringt. TMA befindet sich am Ende des Carnitin-Moleküls, und wird normalerweise durch das in der Leber und auch sonst im Körper zu findende Enzym TMA-Oxidase zu TMAO (Trimethylamin-Oxid) umgebaut, welches selbst wieder geruchlos ist. Bei manchen Menschen liegt nun durch einen genetischen Defekt ein Mangel dieses Enzyms vor, sodass das TMA nicht in ausreichendem Maß zu TMAO verarbeitet werden kann. Diese Menschen leiden am sogenannten “Fischgeruchs-Syndrom”. Dieses liegt bei deinem Kind jedoch mit großer Sicherheit nicht vor. Jedoch kann das Enzym TMA-Oxidase auch einfach durch zu große Mengen an TMA überfordert werden. Wie auch immer − wenn aus dem Darm heraus mehr TMA in den Körper gelangt, als durch das Enzym verarbeitet werden kann, wird es auf anderen Wegen ausgeschieden. Vor allem im Urin und im Schweiß.
Nur 15% des durch den Mund (oral) aufgenommenen Carnitins (z.B. in Form von BioCarn) wird auch als Carnitin vom Körper absorbiert. Der Rest wandert durch den Magen in den Darm und wird dort von bestimmten Bakterien in seine Bausteine aufgespalten. Dabei wird das TMA freigesetzt. Ein Teil davon verbleibt im Darm und führt zum fischigen Geruch in der Windel, ein anderer Teil wird von der Darmschleimhaut aufgenommen und zur Leber transportiert. Wird das dieses TMA zu TMAO verarbeitende Enzym in der Leber durch die Menge an TMA überfordert, gelangt das überschüssige TMA in den Blutkreislauf und wird im gesamten Körper verteilt. Dann ist der fischige Geruch auch in allen Körperausdünstungen enthalten.
Stellt man bei einem Kind auf Carnitintherapie diesen fischigen Geruch fest, ist dies ein deutliches Zeichen dafür, dass mehr Carnitin verabreicht wird, als sein Körper zu verarbeiten in der Lage ist. Erste Maßnahme ist, die tägliche Carnitindosis auf mehrere kleinere Portionen aufzuteilen. Warum das bereits einen deutlichen Unterschied machen kann, lässt sich vielleicht mit dem folgenden Beispiel nachvollziehen: Nehmen wir mal an, du trinkst täglich insgesamt 2 Liter Wasser. Stell dir nun vor, du würdest diese 2 Liter nicht über den Tag hinweg Glas für Glas trinken, sondern am Morgen als eine große Portion zu dir nehmen wollen. Warum auch nicht, schließlich heißt es doch immer nur, dass man “pro Tag” mindestens 2 Liter Wasser trinken soll. Dann kann man es doch auch gleich am Morgen hinter sich bringen. Du kannst dir sicher sehr gut vorstellen, wie das ausgehen wird. Spätestens sobald der Magen voll ist, wird das weiter aufgenommene Wasser zuerst die Speiseröhre und schließlich den Mund füllen und die restlichen 1-1,5 Liter sprudeln dann einfach nur noch aus deinem Mund über dich und machen dich nass. Auf diese Weise hast du also nur 0,5 bis 1 Liter Wasser zu dir genommen und somit dein Tagesziel bei weitem nicht erreicht, denn der ganze Rest ging vollständig verloren. In etwa das Gleiche passiert bei der Einnahme von Carnitin im Körper deines Kindes, wenn die Dosis zu groß ist. Natürlich macht so ein Teelöffel voll Sirup nicht den Magen voll, aber innerhalb einer gewissen Zeitspanne kann halt auch nur eine gewisse (kleine) Menge Carnitin in den Körper aufgenommen werden. Ist die verabreichte Dosis aber z.B. doppelt so groß, geht die zweite Hälfte im übertragenen Sinne daneben und wird dann der oben beschriebenen Verarbeitungskette zugeführt, die mit der Bildung des unangenehmen Fischgeruchs endet. Die Gesamtdosis auf eine halbe Portion morgens und die andere halbe Portion abends aufzuteilen, kann dies unter Umständen bereits wirkungsvoll verhindern. Falls aber selbst das zu keiner Verbesserung führt, sollte in Absprache mit dem Stoffwechselarzt die tägliche Dosis verringert werden.
Der fischige Geruch an sich ist nicht schädlich, aber doch sehr unangenehm − und selbst wenn du dich über kurz oder lang an den “Geruch” deines Kindes gewöhnen und ihn nicht mehr in dem Maße als unangenehm empfinden wirst, nehmen ihn andere Menschen − auch gleichaltrige Kinder im Kindergarten oder in der Schule − doch sehr viel deutlicher wahr. Im Interesse der Gesellschaftsfähigkeit deines Kindes sollte deshalb auch der Stoffwechselarzt bereit sein, geeignete Maßnahmen zur Reduzierung dieses Problems zu ergreifen. Eine Aussage wie “Der Fischgeruch ist ja wohl das kleinere Übel!” ist absolut nicht zu akzeptieren! Fortwährende Hänseleien und gesellschaftliche Ausgrenzung sind für Menschen jeden Alters, aber besonders für Kinder sehr viel schlimmer, als irgend so eine ominöse Stoffwechselstörung, an der sie nach anderer Leute Aussage angeblich leiden sollen, von der sie aber in ihrem normalen Leben üblicherweise überhaupt nichts merken.
Die geringe Absorptionsfähigkeit des oral aufgenommenen Carnitins (die oben erwähnten max 15%) ist Fakt, und es wurde in Studien nachgewiesen, dass es auf diesem Weg grundsätzlich Wochen und Monate dauert, um den körpereigenen Carnitinpool langsam aufzufüllen, egal wie hoch die tägliche Carnitindosis auch sein mag. Je mehr ins Kind reingefüllt wird, desto mehr wird einfach wieder ausgeschieden und desto mehr TMA wird freigesetzt. Daher ist es empfehlenswert in Absprache mit dem Stoffwechselarzt eine auf die tatsächlichen Bedürfnisse des Kindes angepasste Carnitindosis auszutesten, denn die wird seinen Carnitinpool ebenso schnell auffüllen, aber zu weniger unangenehmen Nebenwirkungen führen, denn neben dem fischigen Geruch können zu hohe Carnitindosen auch zu Durchfall und Bauchschmerzen führen.
Sollte die Verordnung der täglichen Carnitingabe übrigens ohne triftigen medizinischen Grund erfolgt sein (in manchen Stoffwechselambulanzen macht man das einfach so, weil man es dort halt so macht), und bisher nichts auf einen sich auch nur ansatzweise abzeichnenden Carnitinmangel hindeuten, wäre ggf. die Einholung einer zweiten Meinung in einer anderen Stoffwechseleinrichtung zu empfehlen.![]() Vor allem dann, wenn bei deinem Kind die häufige bekannte milde MCAD-Variante K329E/Y67H (c.985a>g/c.199t>c) oder eine zu einer milden Form passende gemessene Enzymaktivität von 18% oder höher vorliegt, ist es sehr unwahrscheinlich, dass es zu einem Carnitinmangel kommen könnte.
Vor allem dann, wenn bei deinem Kind die häufige bekannte milde MCAD-Variante K329E/Y67H (c.985a>g/c.199t>c) oder eine zu einer milden Form passende gemessene Enzymaktivität von 18% oder höher vorliegt, ist es sehr unwahrscheinlich, dass es zu einem Carnitinmangel kommen könnte.
Nein, obwohl viele Menschen, die sich erstmals mit dem MCAD-Mangel beschäftigen, eine starke Unterzuckerung (Hypoglykämie) zunächst als alleinige und unter allen Umständen zu verhindernde Hauptgefahr ansehen. Selbst manche Ärzte, die sich noch nicht tiefergehend mit dem Thema befasst haben, sehen darin das eigentliche Problem. Nur so ist jedenfalls zu erklären, dass es bereits mehrere MCAD-Familien gab, die von ihren Stoffwechselärzten ein Blutzuckermessgerät verschrieben bekamen und mit der Anweisung nach Hause geschickt wurden, in Krankheitsphasen (oder generell mehrfach pro Tag) regelmäßig den Blutzuckerwert des Kindes zu messen, und erst in die Klinik zu kommen, wenn er unter einen bestimmten Wert abgefallen wäre. Solange er noch über diesem Wert läge, sei alles bestens, und die Eltern müssten sich um den Zustand ihres Kindes keine Gedanken machen.
Wenn dein Stoffwechselarzt auch zu den so denkenden Menschen gehört, solltest du nach Möglichkeit die Stoffwechselambulanz, oder zumindest den Arzt wechseln. Abgesehen davon, dass diese Blutzucker-Mess-Tortur lediglich dem Kind zerstochene Fingerkuppen und den Eltern eine gewaltigen psychischen Stress beschert, erlaubt die Messung des Blutzuckerwertes keine verlässliche Aussage über den aktuellen Gesundheitszustand des Kindes. Natürlich ist klar: wenn der Blutzuckerwert bei einer Messung tief im Keller ist, kann mit dem Kind etwas nicht stimmen und vermutlich wird man das auch an weiteren körperlichen Symptomen bereits sehen können. Der Umkehrschluss ist jedoch nicht zutreffend: ein trotz längerer Nüchternphase noch normaler Blutzuckerspiegel ist keinesfalls ein sicheres Zeichen dafür, dass mit dem Kind noch alles in Ordnung ist.
Je nachdem, was der Körper des Kindes in einer bestimmten Situation benötigt, wird er hauptsächlich Fett und Glukose oder nur Fett verbrauchen. Kommt es während einer kräfteraubenden sportlichen Betätigung zu einem nahezu vollständigen Verbrauch der Glukose und Glykogenreserven, wird das Gehirn auf die Nutzung von Ketonen umschalten, die immer noch beanspruchten Muskeln verbrauchen jedoch weiterhin Glukose und Fett. Als Folge können in so einer Situation die Glykogenspeicher schnell vollständig verbraucht werden und somit der Blutzuckerspiegel rasch abfallen, was dann auch mit einem BZ-Messgerät deutlich feststellbar wäre. Kommt es dagegen während der Nacht oder im Laufe einer Krankheit mit stark verminderter Nahrungsaufnahme zu einem Energiemangel, wird ebenfalls das Gehirn auf die Nutzung der Ketone umschwenken, jedoch verbrauchen die Muskeln fast keine Energie, und somit auch keine großen Mengen an Glukose. Auch wenn das Kind dann schon MCAD-typische Symptome zeigt, kann der Blutzuckerspiegel trotzdem noch lange auf normalem Niveau bleiben, denn der Körper kann die dann von den Muskeln weiterhin benötigte Glukose durch den Prozess der Glukoneogenese für eine ganze Weile auch noch selbst bilden.
Es empfiehlt sich daher, nicht eine mögliche Hypoglykämie als das alleinige große Problem des MCAD-Mangels zu betrachten, sondern ganz allgemein den bei zu langer Nüchternzeit auftretenden Energiemangel, der sich nicht immer auch am Blutzuckerspiegel zeigt!
Ein Beispiel für lange über den Beginn der Symptome hinaus noch unauffällige Blutzuckerwerte ist in folgender Grafik (Stanley et al, 1990) dargestellt.
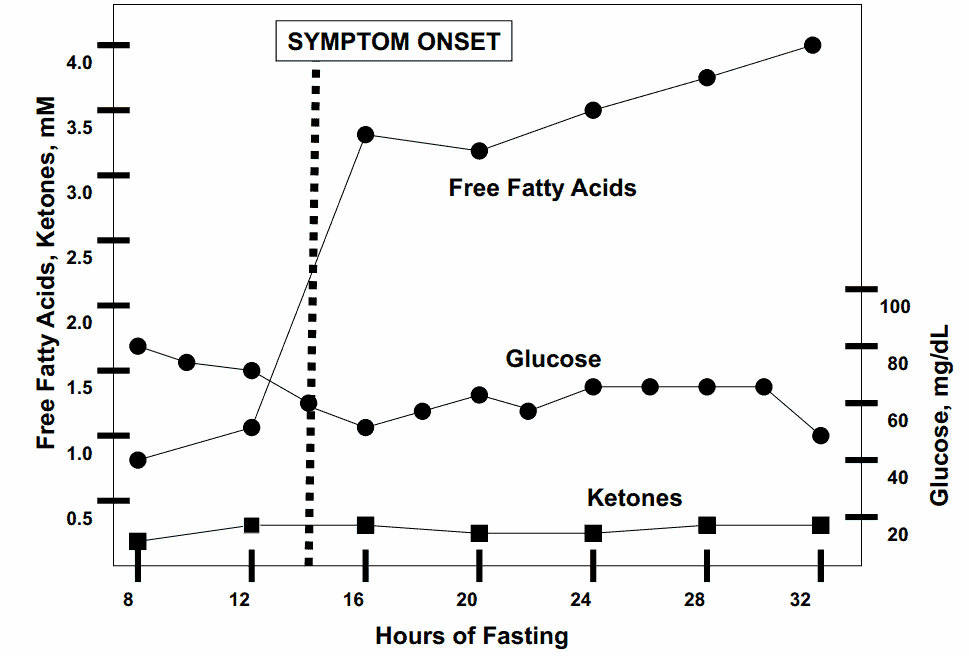
Die Werte der unteren Achse zeigen die Anzahl der Stunden, über die der beobachtete MCAD-Patient nüchtern blieb. Die drei Kurven stellen über die Stunden hinweg den Verlauf des Blutzuckers (Glucose), der im Blut nachweisbaren freien Fettsäuren (Free Fatty Acids) und der aus diesen Fettsäuren in den Mitochondrien der Leber gebildeten und im Blut zum Gehirn transportierten Ketonkörper (Ketones) dar.
Nach einer Nüchternzeit von 12 Stunden (es handelte sich bei dem im Jahr 1990 untersuchten Patienten um einen Jungen nicht näher benannten Alters, dessen MCAD-Mangel zu dem Zeitpunkt noch nicht bekannt war) waren die Glykogenreserven des Patienten soweit aufgebraucht, dass nun die Fettreserven in den Körperspeichern aktiviert wurden. Die Menge der im Blut zu findenden freien Fettsäuren stieg erwartungsgemäß sprunghaft an, was zeigt, dass nun eine bedeutende Menge aus dem Fettgewebe gelöste Fettsäuren durch das Blut zu den verschiedenen Organen (hauptsächlich die Leber) transportiert wurde, um das auftretende Energiedefizit zu kompensieren. Typisch für den MCAD-Mangel ist nun, dass der Verlauf der Ketonkurve nicht dem Anstieg der freien Fettsäuren folgte, wie es bei Menschen ohne MCAD-Mangel der Fall wäre, sondern weiterhin auf seinem niedrigen Niveau blieb. Die direkte Verknüpfung zwischen einerseits der als Energieträger bereitgestellten Fettsäuren und andererseits der daraus gebildeten Ketone ist hier unterbrochen. Genau hierin zeigt sich das Wesen einer Fettsäuren-Oxidations-Störung!
Der gemessene Blutzuckerwert fiel zwar nach den ersten 12 Stunden Nüchternzeit ein wenig ab, blieb aber weiterhin innerhalb eines gewissen Bereichs relativ konstant und man konnte für die gesamten 32 Stunden, während denen der Patient unter Beoachtung stand, keine als solche zu bezeichnende “Hypoglykämie” (Werte unter 50mg/dL) feststellen. Bis einschließlich der Messung in der 30. Stunde war der Blutzuckerwert für einen so lange nüchternen Menschen sogar als durchaus normal zu betrachten.
Trotz dieses die ganze Zeit annähernd normalen Blutzuckerverlaufs zeigten sich bereits ab der 14. Stunde MCAD-typische Symptome (Bewusstseinsstörungen, Apathie, Schläfrigkeit), wie sie auch bei anderen Patienten mit gleichzeitig vorliegender Hypoglykämie beobachtet werden konnten. Besonders das Gehirn, welches zu dem Zeitpunkt auf die Verwertung von Ketonen umschwenkte, war durch den Energiemangelzustand stark unterversorgt.
Im Körper eines Menschen mit MCAD-Mangel passiert nach einer zu langen Nüchternzeit sehr viel mehr, als “nur” eine Unterzuckerung. Eine Entgleisung wirkt sich auf eine ganze Reihe weiterer Vorgänge im menschlichen Körper aus (siehe dazu auch den Artikel “Was ist ein MCAD-Mangel“). Was dabei alles aus der “Schiene springt”, wird in der folgenden Frage behandelt.
![]()
Alle seitens der Stoffwechselexperten genannten Empfehlungen bzgl. maximaler Nüchternzeiten, Carnitingaben, Krankenhausaufenthalte mit Infusionen im Krankheitsfall, usw. verfolgen nur ein Ziel: die im folgenden aufgeführten Entgleisungseffekte zu vermeiden, da diese im Zusammenspiel zu sehr schweren Krankheitsverläufen mit Bewusstlosigkeit, Koma, Organversagen und schließlich auch dem Tod führen können. Diese Symptome können bei Auftreten eines Energiemangels schon lange vor einem per Blutzuckermessgerät feststellbaren Absinkens des Blutzuckerspiegels und einer als solche zu bezeichnenden Hypoglykämie einsetzen.
Auch wenn durch den MCAD-Mangel nur wenige Ketone (siehe vorangegangene Frage) erzeugt werden können, werden doch eine große Menge Fettsäuren aus dem Fettgewebe gelöst und “angeknabbert”. Die übrig bleibenden mittelkettigen Reste werden an Carnitin gekoppelt, und als mittelkettige “Acylcarnitine” aus den Mitochondrien abtransportiert, in den Blutkreislauf abgegeben und somit überallhin transportiert. Diese Acylcarnitine wirken toxisch − besonders auf Gehirnzellen. Je länger so ein Carnitin-Fettsäuren-Molekül ist, desto größer die Toxizität. Die massive Anhäufung mittelkettiger Acylcarnitine führt während einer über längere Zeit nicht gezielt behandelten Entgleisung schließlich zum Koma und bleibenden Gehirnschädigungen.
Ein weiteres Problem mit dem MCAD-Mangel stellt sich jedoch schon etwas früher ein. Bevor die in die Mitochondrien transportierten Fettsäuren überhaupt “angeknabbert” werden können, müssen sie mit einem bestimmten chemischen Stoff, dem Coenzym A (“CoA”) aktiviert werden. Dieses CoA-Molekül wird normalerweise nach getaner Arbeit wieder abgespalten und steht im Körper wieder für eine Vielzahl weiterer chemischer Vorgänge an verschiedenen Stellen zur Verfügung. Beim Abbruch der Fettsäuren-Aufspaltung bei mittelkettiger Länge werden diese Fettsäurenreste mitsamt des zu dem Zeitpunkt noch daran gekoppelten CoAs an Carnitin gebunden und wie zuvor beschrieben als toxisches Acylcarnitin in Umlauf gebracht. Nicht nur das für den Transport durch die Mitochondrienmembran benötigte Carnitin kann auf diese Weise zur Neige gehen, sondern auch das wichtige Coenzym A.
Damit ist es allerdings noch nicht getan. Im menschlichen Körper werden mit der Nahrung aufgenommene, aber nicht direkt benötigte Proteine, im Gegensatz zu Zucker und Fett nicht gespeichert, sondern ausgeschieden. Dies geschieht dadurch, dass sie zunächst in Ammonium (Ammoniak) aufgespalten, und dann über einen größeren Zyklus zu Harnstoff umgewandelt werden. Ammonium selbst ist hochtoxisch für das Gehirn und wird z.B. zur Bildung der Aminosäure Glutamat benötigt, aus der wiederum andere Aminosäuren synthetisiert werden können. Die sich im Körper anstauenden Acylcarnitine stören jedoch bereits den ersten Schritt des Ammonium->Harnstoff-Stoffwechsels, sodass das toxisch wirkende Ammoniak nicht abgebaut wird, sondern sich im Körper ansammelt und zur Anschwellung des Gehirns führt.
An dieser Stelle kann man sich die Frage stellen, woher denn die zu Ammoniak verarbeiteten Proteine kommen sollen, wenn “Nüchternphase” ja gerade bedeutet, dass ein MCAD-Patient überhaupt nichts isst. Auch wenn man über längere Zeit keine Nahrung zu sich nimmt, hat der Körper nach einigen Stunden wieder neuen Bedarf an Proteinen, da diese, wie zuvor erwähnt, nicht gespeichert werden. Zu diesem Zweck holt sich der Körper dann einfach die Proteine von den Stellen, an denen sie noch vorhanden sind. Er baut Muskeln ab und transportiert die frei werdenden Proteine zu den Stellen, an denen sie benötigt werden. Während einer laufenden Entgleisung erfolgt dieser Muskelabbau jedoch nicht an den tatsächlichen Bedarf angepasst, sondern völlig unkontrolliert. Es kommt zu einem regelrechten Muskelzerfall, und die dabei freigesetzten viel zu großen Mengen an Proteinen werden wiederum zu Ammoniak umgewandelt. Der Muskelzerfall (Rhabdomyolyse) erstreckt sich vor allem auf die “quergestreiften” Muskelfasern, wie sie in der Skelettmuskulatur, dem Herzen und dem Zwerchfell vorliegen. Ein schon deutlich fortgeschrittener Muskelzerfall zeigt sich z.B. an einer Braunfärbung des Urins, die von der Freisetzung des in den zersetzten Muskelfasern vorhandenen Farbstoffs Myoglobin herrührt.
Darüber hinaus stören die Acylcarnitine den ohnehin schon aufgrund des MCAD-Mangels eingeschränkten Fettstoffwechsel in der Leber. Dieser zusätzlichen Beeinträchtigung der normalen Leberfunktion versucht der Körper durch verstärkte Ausschüttung von Leber-Enzymen entgegenzuwirken. Diese bewirken einen ständig steigenden Transport von Fettsäuren in die Leber hinein, sodass es innerhalb weniger Stunden zu einer massiven Leberverfettung kommt. Diese ist jedoch reversibel, wenn rechtzeitig die notwendigen Gegenmaßnahmen getroffen werden.
Alles das zusammen macht die Gefährlichkeit einer Stoffwechselentgleisung aus: Der Energiemangel an sich, die Anreicherung toxischer Stoffwechselzwischenprodukte, der sich möglicherweise noch dazu einstellende Carnitinmangel, der übermäßige Verbrauch von Coenzym A, durch den weitere Enzyme und Stoffwechselvorgänge im Bereich der Glukose-Erzeugung beeinträchtigt werden, die Ansammlung des giftigen Ammoniaks, der dies begleitende Muskelzerfall und die rasante Verfettung der Leber.
Das klingt alles jetzt sehr dramatisch, und genauso ist es auch! Die gute Nachricht ist, dass es aufgrund der frühzeitigen Information der Eltern über den bei ihrem Kind vorliegenden MCAD-Mangel in den letzten Jahren zu fast gar keinen auch nur annähernd so schweren Stoffwechselkrisen mehr gekommen ist. Die aufgezählten schlimmen Folgen werden aber nicht durch regelmäßige Blutzuckermessungen verhindert, sondern dadurch, dass die Eltern beim ersten Auftreten offensichtlicher Symptome möglichst schnell den Notarzt verständigten, und sich nicht erst noch auf die nutzlose Suche nach dem Blutzuckermessgerät machen, um sich durch vermeintlich noch gute Werte viel zu lange in Sicherheit zu wiegen.
Die einfachste und zugleich wirksamste Maßnahme, um ein Auftreten der Symptome im Vorfeld zu verhindern, besteht darin, darauf zu achten, dass das Kind, der Jugendliche, der Erwachsene mit MCAD-Mangel niemals zu lange nüchtern ist, sondern regelmäßig Nahrung zu sich nimmt, um nie in einen Energiemangelzustand zu geraten. Dies schließt auch vorsorgliche Krankenhausaufenthalte zwecks Infusionen mit ein.![]()
Sicher hast du bereits von einem Arzt oder auch beim Lesen dieser Seiten erfahren, dass die wichtigste Maßnahme zur Verhinderung der früher oft schwerwiegenden Auswirkungen des MCAD-Mangels darin besteht, die Mahlzeitenabstände nicht zu groß werden zu lassen.
Die folgende Tabelle zeigt ein mögliches Beispiel der empfohlenen maximalen Nüchternzeiten für Kinder unterschiedlichen Alters.
Falls du von deinem Stoffwechselarzt die MCAD-Broschüre von Nutricia Metabolics (früher von Milupa) ausgehändigt bekommen hast, unterscheiden sich im Vergleich dazu sowohl die aufgeführten Altersbereiche, als auch die angegebenen maximalen Nüchternzeiten. Eventuell hast du aber auch eine von deiner Stoffwechselambulanz selbst zusammengestellte Tabelle erhalten, die wiederum andere Altersbereiche und Zeitabstände nennt.
In den damaligen Forumsdiskussionen zeigte sich immer wieder, dass sich aufgrund der Verwirrung über die unterschiedlichen Tabellen und überhaupt wegen der teilweise stark voneinander abweichenden Nüchternzeiten bei den Eltern eine ganze Reihe von Missverständnissen festsetzten, die nach einiger Zeit nur noch schwer wieder überwunden werden konnten. Teilweise wurden und werden diese Missverständnisse sogar durch die betreuenden Stoffwechselärzte selbst hervorgerufen, da diese oft selbst ein etwas verzerrtes Bild von der Bedeutung dieser Tabellen haben. Aus diesem Grund soll an dieser Stelle auf die gravierendsten Missverständnisse eingegangen werden.
Nein, sondern es handelt sich um maximale Zeitabstände, die nach Belieben unterschritten werden können, aber nach Möglichkeit nicht überschritten werden sollten. Leider gibt es in fast jedem Bekannten- oder auch Freundeskreis immer wieder andere “wohlmeinende” Mütter, die sich mit Kommentaren wie “Na, mit 6 Monaten muss ein Kind ja wohl in der Lage sein, schon mal 8 Stunden am Stück zu schlafen! Meines konnte das jedenfalls!” nicht zurückhalten können. Diese Einstellung solltest du im Interesse deines Kindes nicht für dich selbst übernehmen. Auch wenn laut Tabelle für Säuglinge >6 Monate 8 Stunden Nüchternzeit durchaus zulässig zu sein scheinen, bedeutet dies nicht, dass ein Kind dann auch darauf trainiert werden muss, diese 8 Stunden tatsächlich nüchtern zu bleiben. Sollte dein Kind zu den wenigen Exemplaren gehören, die tatsächlich schon mit 6 Monaten 8 und mehr Stunden am Stück und ohne zwischenzeitliche Nahrungsaufnahme durchschlafen wollen, bedeuten diese Zeiten nur, dass es spätestens nach diesen 8 Stunden von dir geweckt werden sollte, um ihm neue Nahrung zuzuführen.
Da die Tabelle in der alten Milupa-Broschüre pro Altersbereich nur eine zeitliche Angabe aufführt, kann dieser Eindruck wirklich entstehen − vor allem dann, wenn man den Textabschnitt, in den die Tabelle eingebettet ist, nicht sorgfältig liest. Daraus geht nämlich hervor, dass die in der Tabelle genannten Zeiten sich auf die nicht zu überschreitenden nächtlichen Nahrungspausen beziehen.
Zwischen Abendessen und Frühstück liegt im Normalfall ein deutlich größerer Abstand (>12 Stunden), als zwischen Frühstück und Mittagessen oder zwischen Mittag- und Abendessen, wobei die Pausen zwischen diesen Mahlzeiten meist ohnehin noch durch Zwischenmahlzeiten wie Obst oder andere Snacks ausgefüllt werden. All dies ist aber während des Nachtschlafs nicht gegeben. Daher weist die frühere Milupa-Broschüre mit dieser Tabelle ausdrücklich darauf hin, dass insbesondere nachts die angegebenen Zeiten nicht überschritten werden sollten. Es ist deshalb angeraten, eine Spät- oder sogar Nachtmahlzeit zur Sicherheit von Anfang an als festen Bestandteil jeder Nacht einzurichten.
Auf die tagsüber im Auge zu behaltenden maximalen Nüchternzeiten geht die alte Milupa-Broschüre jedoch überhaupt nicht ein, da man wohl annahm, dass kritische Energiemangelzustände tagsüber aufgrund der Haupt- und Zwischenmahlzeiten ohnehin nicht eintreten werden und die Eltern vor allem auch sehr schnell merken werden, wenn das Kind apathisch wirkt oder sich sonstwie eigenartig verhält. Sie sind dann in der Lage entsprechend mit neuer Nahrung gegensteuern zu können. Während des Nachtschlafs sieht man dem Kind dagegen nicht an, wenn es in einen kritischen Energiemangelzustand abgleitet. Deshalb legt die Broschüre mit dieser Tabelle den besonderen Augenmerk auf die nach Möglichkeit nicht zu überschreitenden und für das jeweilige Alter als sicher geltenden nächtlichen maximalen Nüchternzeiten.
Es wird aber jedem einsichtig sein, dass ein Kind, Jugendlicher und auch Erwachsener während des Tages und somit der wachen und körperlich aktiven Zeit deutlich mehr Energie verbraucht, als während des Nachtschlafs, wenn alle Körperfunktionen auf Sparflamme laufen und der gesamte Anteil des Leistungsumsatzes (siehe dazu diesen Artikel) wegfällt. Wenn die alte Milupa-Tabelle somit die maximalen Nüchternzeiten während der Nacht nennt (und genau darauf weist sie ausdrücklich hin), müssen die für tagsüber anzusetzenden Zeiten dem hinzukommenden Energiebedarf aufgrund des Leistungsumsatzes Rechnung tragen und folglich etwas verkürzt werden (siehe die obige Tabelle).
Die Tabelle der Milupa-Broschüre enthält deshalb nicht, wie es von vielen Eltern und auch Ärzten verstanden wird, eine deutlich großzügigere Regelung für die während des Tages im Blick zu behaltenden Nüchternzeiten, sondern sie unterlässt es einfach nur, hierzu eine deutliche Aussage zu machen.
Aus dem oben formulierten 1. Missverständnis in Kombination mit dem Vorliegen der alten Milupa-Broschüre resultiert oft auch eine große Unsicherheit − nämlich genau dann, wenn entweder der Tag erreicht ist, an dem das Kind laut Tabelle von dem einen Altersbereich in den nächsten rutscht, oder wenn man bei einem der regelmäßigen Kontrolltermine in der Stoffwechselambulanz mitgeteilt bekommt, dass das Kind aufgrund seines Alters jetzt plötzlich nachts zwei Stunden länger schlafen kann.
Lass dir nichts einreden! Dein Kind könnte laut Aussage der Tabelle nun vermutlich bedenkenlos bis zu zwei Stunden länger schlafen, aber es muss nicht! Ganz oft schildern betroffene Eltern, dass für ihr Kind jetzt eine um zwei Stunden längere Nüchternzeit “gelte”, sie sich mit dem Gedanken aber nicht so richtig anfreunden könnten, sondern lieber bei den bis jetzt geltenden Zeiten bleiben würden, weil sie sich damit inzwischen gut und sicher fühlen. Manche dieser Eltern stellen die nächtliche Weckzeit dann aber auch wirklich von heute auf morgen um zwei Stunden vor, und haben wochen- und monatelang ein schlechtes Gefühl dabei; andere wiederum erhöhen den zeitlichen Abstand erst einmal nur um eine halbe Stunde oder auch gar nicht, und haben dabei ebenfalls ein schlechtes Gewissen − weil es der Arzt doch anders angeordnet hatte!
Beide Haltungen resultieren aus einem falschen Verständnis über die Bedeutung und die Verbindlichkeit der in den Tabellen genannten Zeiten, und dieses falsche Verständnis gilt es frühzeitig abzulegen!
Die in den Tabellen oder vom Stoffwechselarzt genannten Zeiten sind lediglich Empfehlungen, keine verbindlichen Regeln. In deiner Rolle als Rat suchendes Elternteil hast du den Arzt gefragt, was du beachten musst, damit deinem Kind nichts passiert, und der Arzt hat dir daraufhin eine Tabelle oder eine Stundenangabe an die Hand gegeben, bei deren Nichtüberschreitung es der Erfahrung der letzten Jahre nach zu keinen Stoffwechselentgleisungen gekommen ist. Mehr ist es nicht. Die Tabelle, bzw. die darin enthaltenen Zeiten spiegeln Erfahrungswerte und somit Empfehlungen einzelner Ärzte oder Kliniken wieder (halt derjenigen, die eine entsprechende Tabelle letztlich veröffentlicht haben), die dir lediglich helfen sollen, ein Gefühl für den sicheren Umgang mit dem MCAD-Mangel zu bekommen. Es handelt sich nicht um Anweisungen, die es zu befolgen gilt, und bei deren Nichtbefolgung du mit berechtigtem Tadel durch deinen Arzt, oder sogar mit rechtlichen Konsequenzen zu rechnen hättest. Insofern ist es völlig dir überlassen, ob du die für das Alter deines Kindes laut Erfahrungen der Ärzte problemlos sicheren Zeiten bis zum Ende ausreizt, oder ob du sie “eigenmächtig” um ein oder mehr Stunden verkürzt. Sogar verlängern könntest du die Zeiten − aber wer macht das schon?
Es kann großen Sinn machen, das Kind von Anfang an langsam an eine immer zur genau gleichen Zeit erfolgende nächtliche Zwischenmahlzeit zur Auffrischung der Kohlenhydratvorräte zu gewöhnen, z.B. um 2 Uhr nachts. Es wäre völlig unsinnig, diese Nachtmahlzeit irgendwann auf 4 Uhr zu verschieben, bloß weil das Kind laut Tabelle nun 2 Stunden mehr abkönnen müsste, oder weil das Kind sein Abendessen inzwischen immer zu einem späteren Zeitpunkt zu sich nimmt. Zeitliche Verschiebungen würden sowohl bei deinem Kind als auch bei dir zu unnötigem Stress und schlechtem Schlaf führen. Führe lieber ein Ritual bzw. einen festen Zeitpunkt für die Nachtmahlzeit ein, an dem dein Kind auch in späteren Jahren noch festhalten kann − selbst wenn der zeitliche Abstand zum Abendessen dann nur 6 Stunden beträgt, und dein Kind laut Tabelle schon 10 Stunden verkraften könnte.
Besonders groß sind die Unterschiede zwischen den kursierenden Tabellen ja überhaupt nicht, aber auch kleine Details können in der Praxis zu großen Problemen führen. Stell dir dich selbst in der folgenden Situation vor: laut Aussage deines Stoffwechselarztes solltest du dein Kind bis zum Alter von 6 Monaten auf keinen Fall länger als 6 Stunden nüchtern lassen. Du hast also den Wecker nachts nach dem letzten Füttern immer um 6 Stunden weiter gestellt. Nach diesen 6 Monaten bekommst du beim neuen Termin in der Stoffwechselambulanz vom Arzt gesagt, dass dein Kind ab jetzt immer bis zu 8 Stunden nüchtern bleiben könne. Was hat sich von gestern auf heute geändert? Das Kind ist einen Tag älter geworden, sonst nichts! Es darf aber plötzlich zwei Stunden länger nüchtern bleiben. Warum durfte es das dann gestern noch nicht? Und warum “gelten” diese 8 Stunden dann laut Milupa-Broschüre bis zum Ende des dritten Lebensjahres − und dann sind es bis zum Ende des 7. Lebensjahres maximal 10 Stunden. Was verändert sich bei dem Kind am Tag des vierten Geburtstags, sodass es da wieder zwei Stunden mehr sein dürfen.
Von einem Tag zum anderen sind die bei deinem Kind stattfindenden körperlichen Veränderungen natürlich sehr gering und kaum wahrnehmbar. Die Tabelle der alten Milupa-Broschüre fasst deshalb zur reinen Vereinfachung die verschiedenen Altersabschnitte zusammen und nennt als maximale Nüchterntoleranzzeit einen Mittelwert. Ob dieser Mittelwert aber am Anfang des Alterszeitraumes, an seiner Mitte oder am Ende ausgerichtet wurde, ist aus der Tabelle jedoch nicht zu ersehen. Wenn die angegebenen Zeiten, so wie es im Text klingt, tatsächlich die maximale zeitliche Ausbaustufe für einen bestimmten Altersbereich darstellen, ist es schwer verständlich, weshalb dann plötzlich von heute auf morgen zwei weitere Stunden Nüchternzeit in Ordnung sein sollten.
Die obige Tabelle drückt den tatsächlichen Sachverhalt etwas besser aus. Zusätzlich zu den ergänzend aufgenommenen Empfehlungen für Neugeborene (die ersten 3-4 Wochen) und Säuglinge innerhalb der ersten 3 Monate, für die 6 Stunden Nüchternzeit alleine schon vom persönlichen Gefühl her viel zu lang erscheinen, sind auch für die restlichen Altersbereiche keine festen Stundenangaben, sondern Zeitbereiche genannt. Diese sollen verdeutlichen, dass es z.B. für einen Säugling ab 3 Monate einfach mit den zuvor als maximale Grenze empfohlenen 4 Stunden weitergehen kann und die Eltern diese Zeitspanne bis zum Ende des 6. Monates allmählich auf bis zu 6 Stunden ausdehnen können. Zu Beginn des nächsten Altersbereichs (6-12 Monate) geht es mit diesen 6 Stunden weiter, und wenn Eltern und Kind dazu bereit sind, können sie nach eigenem Tempo die Zeiten bis zum ersten Geburtstag langsam auf bis zu 8 Stunden strecken − müssen sie aber nicht. Diese Tabelle drückt somit etwas besser die eigentlich lineare Steigerungsmöglichkeit der maximalen Nüchternzeiten aus, als die Milupa-Tabelle mit den abrupten 2-Stunden-Sprüngen.
Zusätzlich verdeutlicht diese Tabelle noch, dass die als wirklich wichtigste Maßnahme im Auge zu behaltenden maximalen Nüchternzeiten nicht mit dem Erreichen des Schulalters bedeutungslos werden, sondern das ganze Leben lang beibehalten werden müssen, um immer auf der sicheren Seite zu sein.
Dieses Missverständnis resultiert manchmal aus dem eigentlich korrekten Verständnis der 1. Aussage − nämlich, dass es sich bei den in der Tabelle angegebenen Zeiten nicht um einzuhaltende Mahlzeitenabstände, sondern um maximale Nüchternzeiten handelt. Der Begriff “maximale” erweckt dabei aber den Eindruck, dass es sich dabei um die alleräußerste Grenze handelt und die Kinder selbst bei ganz kurzen Überschreitungen dieser Zeiten sofort in eine riskante Stoffwechselkrise abrutschen könnten. Es scheint, dass diese auf das jeweilige Alter bezogenen Zeiten immer den am Rande der Klippe entlang des Abgrundes führenden Weg beschreiben würden und die kleinste Abweichung zur falschen Seite sofort den Absturz nach sich ziehen könnte.
Vermutlich jedes Elternpaar erlebt am Anfang seines Weges mit dem MCAD-Mangel die Situation, dass man nachts einmal aufgrund eigener Erschöpfung den für das rechtzeitige Füttern des Kindes gestellten Wecker einfach wieder abschaltet und sofort erneut einschläft. Irgendwann wacht man dann mit einem riesigen Schrecken auf, steht förmlich im Bett, blickt ungläubig auf die Uhrzeit und realisiert voller Panik, dass man die “zulässige” maximale Nüchternzeit um ein bis mehrere Stunden überschritten hat. Man rennt zum Bettchen des Kindes, voller Angst, dass es aufgrund dieses einmaligen Versehens bereits verstorben sein könnte.
Diese Angst ist aber eigentlich unnötig. Obwohl man den MCAD-Mangel nicht auf die leichte Schulter nehmen und deshalb die Nichtüberschreitung der empfohlenen Zeitabstände immer im Blick behalten sollte, enthalten diese Zeiten, bezogen auf ein gerade rundum gesundes Kind, einen sehr großen Sicherheitspuffer. Man bewegt sich mit einer maximalen Nüchterntoleranzzeit von z.B. 8 Stunden also im Normalfall nicht direkt am Rand der Klippe, sondern auf einem Weg, der mehrere Meter vom Abgrund entfernt entlangführt.
Die ganzen Zeitempfehlungen basieren auf statistischen Auswertungen von einzelnen an unterschiedlichen Kliniken durchgeführten Fastenversuchen, sowie aus der medizinischen Fachliteratur zusammengetragenen Berichten von festgestellten Entgleisungen der vergangenen Jahre (größtenteils aus der Zeit vor dem Neugeborenenscreening) und den daraus ermittelten Ergebnissen für sichere und unsichere Zeiten.
Als sichere Zeiten betrachtet man diejenigen Wertepaare (Alter des Kindes / Nüchternzeit), bei denen bis zum erneuten Füttern keinerlei Symptome oder deutlich sinkende Blutzuckerwerte aufgetreten sind. Die dabei gemessenen Zeiten zwischen 8 und 24 Stunden bedeuten aber nicht, dass es bei den betreffenden Kindern nur bis zu dem jeweiligen Zeitpunkt gut lief, und der Stoffwechsel unmittelbar danach gekippt wäre. Manche dieser Fastenversuche waren von Beginn an für eine bestimmte Zeitspanne angesetzt, manche wurden auch nach einer gewissen Zeit von den Eltern abgebrochen, weil man den nach Nahrung verlangenden Säugling oder das Kleinkind einfach nicht mehr länger im Dienste der Wissenschaft hungern lassen wollte. Die in den Versuchen ermittelten sicheren Zeiten bedeuten somit ganz einfach nur: Das Kind hat während des Experiments für eine bestimmte Zeit keine Nahrung zu sich genommen, und es ist ihm nichts passiert.
Für die Aufstellung von Empfehlungstabellen zu maximalen Nüchterntoleranzzeiten sind diese teilweise sehr langen sicheren Zeiten aber relativ belanglos, denn an anderen Tagen, mit anderer Zusammensetzung der vorangegangenen Mahlzeit oder anderem Gesundheitszustand könnte das Ergebnis bei diesen Kindern auch anders ausgesehen haben. Nützlich sind daher vor allem die Ergebnisse aus Versuchen oder in der Literatur berichteten Entgleisungsfällen, bei denen sich nach einer gewissen Zeit dann doch erste im Verhalten des Kindes erkennbare Energiemangelsymptome oder ein abfallender Blutzuckerspiegel zeigten. Diese Symptome traten den Studien zufolge bei den untersuchten Kindern bis 30 Monate hauptsächlich im Bereich zwischen 13 bis 20 Stunden auf, es gab aber auch Ausnahmen, bei denen erst jenseits der 30 Stunden erste Auffälligkeiten festgestellt wurden. Ziel der Empfehlungstabellen ist es jedoch, mit der auf einen bestimmten Altersbereich bezogenen maximalen Nüchterntoleranzzeit grundsätzlich deutlich unter der kürzesten dabei gemessenen Zeitspanne zu bleiben. Da es weltweit nur wenige dieser Fastentests gab, kann aus den daher auch nur wenigen statistisch auswertbaren Daten nicht sicher geschlussfolgert werden, dass 13 Stunden für ein Alter von 12 Monaten tatsächlich die unterste Grenze sind und es nicht bei einem anderen, nicht untersuchten Kind vielleicht auch schon nach 11 oder 12 Stunden zu Symptomen gekommen wäre. Daher liegen die empfohlenen maximalen Nüchterntoleranzzeiten je Altersbereich immer deutlich − also mehrere Stunden − unterhalb der kürzesten bisher getesteten Zeitspanne, ab der erste Symptome aufgetreten sind.
An dieser Stelle soll aber auch nochmal auf die Bedeutung der zuvor beschriebenen sicheren Zeiten zurückgekommen werden. Auch wenn die verschiedenen kursierenden Tabellen der maximalen Nüchterntoleranzzeiten es suggerieren − es gibt keine harte Grenze zwischen sicheren und unsicheren Zeiten. Bei den meisten Säuglingen und Kleinkindern sind in diesen überwachten Versuchen auch lange nach Überschreitung der üblichen empfohlenen Zeiten noch keinerlei Symptome aufgetreten.
Diese letzte Feststellung soll aber keineswegs zur Unachtsamkeit oder gar absichtlichen Ausdehnung der Mahlzeitenabstände über die empfohlenen Zeiten hinweg ermuntern. Sie soll lediglich zur Beruhigung beitragen, dass sich das eigene Kind bei Nichtüberschreitung der maximalen Nüchterntoleranzzeiten − insbesondere in gesunden Zeiten, also ohne Vorliegen einer Krankheit mit erhöhtem Energiebedarf oder Nahrungsverweigerung − sehr weit im sicheren Bereich befindet und kein Grund zur Sorge besteht.
Manche Eltern empfinden die in den Tabellen genannten Zeiten für die maximalen Nüchterntoleranzzeiten über Jahre hinweg als warnend erhobenen Zeigefinger, als von den Ärzten auferlegt Vorschrift für das tägliche Leben oder gar als ständig präsente Bedrohung (“wenn ich auch nur einmal unachtsam bin und die Zeiten überschreite, passiert meinem Kind was − denn jenseits der x Stunden befindet sich der Abgrund.”)
Diese Sichtweise basiert jedoch auf möglicherweise vorliegenden Missverständnissen, wie oben aufgezeigt, denn diese Tabellen sollen nicht dazu dienen, die Eltern und später die Kinder selbst mit Vorschriften zu knechten, sondern ihnen einen möglichst unbeschwerten Umgang mit dem MCAD-Mangel im täglichen Leben ermöglichen. Sie sind nicht als “wenn du das nicht penibel einhältst, passiert was Schlimmes!” zu verstehen, sondern als “wenn du dein Kind einfach nur innerhalb der Zeiten dieser Tabelle fütterst, ist es nach allen bisherigen Erfahrungen völlig sicher. Mach dir keinen überflüssigen Stress.” ![]()
Diese Frage wird mir oft von verschiedenen Teilnehmern der Facebook-Gruppe gestellt, weil dort anscheinend annähernd regelmäßig die Frage angesprochen wird, welche Nüchternzeiten für andere Kinder eines bestimmten Alters gelten. Auch im früheren Forum tauchte die Zeiten-Frage mit schöner Regelmäßigkeit auf, und nicht nur von ganz neu betroffenen Eltern. Mit jedem neuen Lebensjahr der Kinder schien es wieder aufs neue wichtig zu sein, einmal abzufragen, wie denn die Empfehlungen der anderen Stoffwechselambulanzen nun wieder aussähen. Ich hätte mir vielleicht mal die Mühe machen sollen, eine Tabelle für die von den verschiedenen Kliniken verteilten Empfehlungslisten zu erstellen, in der dann zeilenweise leicht erkennbar gewesen wäre, wie viele Stunden für die einzelnen Altersgruppen genannt werden. Aber ich glaube, selbst dann wäre diese Frage wieder und wieder gestellt worden. Das Interesse daran, ob man im Vergleich mit den an anderen SWAs betreuten Familien diesbezüglich besser oder schlechter gestellt ist, bzw. ob man die Empfehlungen des eigenen Arztes bestätigt sehen kann oder ob es in Einzelfällen vielleicht doch mal individuell an den Kindern ausgerichtete Zeiten gibt, ist einfach nicht zu unterdrücken.
Hintergrund war und ist meistens, dass die eigenen maximalen Nüchternzeiten noch so bemessen sind, dass man um eine Spät- oder sogar Nachtmahlzeit nicht herumkommt, diese aber gerne so langsam einstellen würde und deshalb herumfragt, wie das an anderen Stoffwechselambulanzen gehandhabt wird. Mit einer seitens der Klinik empfohlenen Nüchternzeit von 12 Stunden wäre das Auslassen einer zusätzlichen Mahlzeit schon drin, 10 Stunden würden nicht ganz ausreichen und mit sogar nur 8 Stunden wäre es vollkommen unmöglich. Im Vergleich zeigt sich dann, dass es Stoffwechselambulanzen gibt, deren Empfehlungstabellen bereits zweijährigen Kinder 12 Stunden in der Nacht zugestehen, während andere sogar während der ersten paar Grundschuljahre immer noch maximal 10 Stunden, teilweise sogar nur 8 Stunden empfehlen.
Woran liegt das? Wird mit dieser Bandbreite der Zeiten dem Umstand Rechnung getragen, dass Kinder nun mal unterschiedlich sind und dass deshalb für manche etwas kleiner oder sogar schwächlicher wirkenden Kinder kürze Zeiten und für besonders fit und wohlgenährt erscheinende Kinder dann gleich die vollen 12 Stunden zugestanden werden? Ganz und gar nicht! Die in den von einer Klinik an die MCAD-Eltern verteilten Tabellen genannten Zeiten sind für alle dort betreuten Kinder gleich. Um auf die individuellen Bedürfnisse eines Kindes einzugehen (schlechter Esser/guter Esser) wird den Eltern lediglich dazu gesagt, dass sie immer dann, wenn sie “das Gefühl haben”, dass die aufgenommene Energiemenge vielleicht etwas zu gering sein könnte, einfach mal etwas früher die nächste Mahlzeit anbieten. Im Klartext bedeutet dieses “wenn Sie das Gefühl haben” nichts anderes, als: “Wir als Stoffwechselambulanz haben hier so ‘ne Tabelle mit Zeiten, die wir vor einigen Jahren mal in irgendeinem Buch oder Artikel gefunden und abgeschrieben haben. Wir empfehlen ihnen, sich danach zu richten, aber letztendlich liegt die Verantwortung voll und ganz bei ihnen als Eltern. Wenn ihr Kind trotz Einhaltung der Zeiten in einen Energiemangel rutscht, hätte doch wohl vorher auffallen müssen, dass ihr Kind nicht genug zu sich genommen hat oder irgendeine Krankheit ausbrütet! Sie hätten ihm dann einfach in kürzeren Abständen Nahrung anbieten müssen”.
Das klingt jetzt vielleicht hart, ist aber genau so gemeint. Sollte einem Kind trotz Einhaltung der Zeiten etwas passieren, wird keine einzige Klinik dafür die Verantwortung übernehmen! Dann wird deutlich herausgestellt, dass die Tabelle nur gut gemeinte Empfehlungen und keinerlei ärztliche Anweisungen enthält.
Möge diese Situation niemals eintreten, aber wenn einem erstmal deutlich wird, welche Bedeutung die Stoffwechselambulanzen selbst der Verbindlichkeit dieser Zeiten im Ernstfall einräumen werden, zeigt sich um so deutlicher, dass die Verantwortung und deshalb auch die alleinige Entscheidungsgewalt bzgl. solcher Zeiten voll und ganz bei den Eltern liegt.
Die euch vorliegende Tabelle wurde aus der Schublade gezogen, weil man euch als noch unerfahrenen Eltern etwas an die Hand geben wollte, damit ihr überhaupt erstmal ein Gefühl dafür entwickeln könnt, wie regelmäßig euer Kind über die nächsten paar Jahre hinweg (in denen es noch nicht in der Lage ist, selbst auf die Signale seines Körpers zu achten) Nahrung zu sich nehmen sollte. Solange die Eltern in den ersten Monaten und manchmal auch Jahren noch voller Sorgen und Ängsten sind, ist so eine Tabelle mit “vermeintlich ärztlichen Anweisungen” tatsächlich etwas sehr Beruhigendes, weil es das Gefühl vermittelt, zumindest den Punkt mit den maximalen Mahlzeiten-Abständen nicht alleine entscheiden zu müssen.
Kommen wir aber zur eigentlichen Frage zurück. Warum sind es z.B. bei eurem Kind mit 4 Jahren noch 8 Stunden und bei einem Kind im gleichen Alter 10 und bei wieder einem anderen Kind sogar 12 Stunden? Einfach nur, weil ihr in der Uniklinik A betreut werdet, Kind 2 in Uniklinik B und Kind 3 in Uniklinik C. Würdet ihr näher an Uniklinik C wohnen und dort betreut werden und Kind 3 näher an Klinik A, hättet ihr jetzt schon die 12 Stunden auf dem Papier stehen und Kind 3 wäre erst bei 8. Würdet ihr dann denken, dass 12 Stunden für euer Kind doch ein bisschen sehr lang sein könnten, weil man den in Klinik A betreuten Kindern nur 8 Stunden zugesteht? Höchstwahrscheinlich nicht! Würden die Eltern von Kind 3 (mit jetzt nur 8 statt 12 Stunden) ihrerseits in Erwägung ziehen, dass sie die nächtliche Nüchternzeit auch genauso gut auf 12 Stunden ausdehnen könnten, weil sie das Kind aufgrund der 8 Stunden jede Nacht nochmal extra wecken müssen und es in anderen Kliniken ja auch geht? Sie würden es sich vermutlich wünschen, das von ihrem Stoffwechselarzt “genehmigt” zu bekommen, aber in ihrer getauschten SWA in Klinik A schaltet man auf stur und stellt ihnen ggü. klar, dass dies auf keinen Fall ginge! Acht Stunden seien momentan noch das höchste der Gefühle und wenn man das woanders anders sähe, hätten die dort einfach nur keine Ahnung und würden die Kinder einem viel zu hohen Risiko aussetzen!
An diesem kleinen Tausch-Gedankenexperiment wird hoffentlich klar, dass es nur dem Zufall des jeweiligen Wohnortes geschuldet ist, dass Kinder gleichen Alters völlig unterschiedliche maximale Nüchternzeiten auf dem Papier stehen haben. Würden jetzt Kinder aus den SWA, in denen 12 Stunden in der Tabelle stehen, häufiger in eine Stoffwechselkrise geraten, als die Kinder aus den SWA mit 10 oder nur 8 Stunden, könnte man daraus ableiten, dass die 12 Stunden doch etwas zu hoch gegriffen sein könnten und die niedrigeren Zeiten deshalb “besser” seien. Das scheint aber nicht der Fall zu sein.
Deshalb kannst du genaugenommen machen, was du willst. Es liegt alles letztlich in deiner alleinigen Verantwortung, denn wie oben eingangs erläutert, wird auch seitens eurer SWA keinerlei Garantie für die Unbedenklichkeit der in ihrer jeweiligen Tabelle stehenden Zeiten übernommen werden. Es seid immer ihr als Eltern, die in jeder Situation nach bestem Wissen, Gewissen und mit ganz viel Bauchgefühl entscheiden müsst, wie ihr mit eurem Kind umgehen sollt.
Soweit zur Theorie! Rein praktisch werdet ihr gegenüber eurem Stoffwechselarzt mit der Anfrage, ob ihr nicht auch 12 Stunden Nüchternzeit in der Nacht einführen könntet, mit sehr großer Wahrscheinlichkeit auf taube Ohren stoßen. Wie allen Ärzten wird auch ihm oder ihr völlig egal sein, was man anderswo macht, denn HIER empfiehlt man es nun mal so. Wenn sich Eltern nicht daran halten wollen, sei das ganz alleine ihre Verantwortung, wenn dem Kind dann was Schlimmes passiere! Na gut – das ist es sowieso, also was soll’s? So locker über den Dingen zu stehen und die Meinung des Arztes Meinung des Arztes sein zu lassen, ist aber natürlich nicht leicht, denn schließlich möchte man ja schon ein gutes Arzt-Patienten-Verhältnis beibehalten.
Deshalb hast du im Prinzip drei Möglichkeiten:
Welche dieser Möglichkeiten ich dir empfehlen würde? Verrate ich nicht! Mach doch, was DU willst!
![]()
Als ich Anfang 2008 das MCAD-Forum ins Leben rief, stellte sich bereits innerhalb weniger Wochen sehr deutlich die ziemlich verwirrende, wenn nicht gar verstörende Erkenntnis ein, dass die an verschiedenen Stoffwechselambulanzen betreuten Familien von ganz unterschiedlichen Vorgehensweisen bzw. Empfehlungen ihrer Ärzte berichteten. Dies betraf sowohl die ihnen gegenüber gemachten Aussagen zu Nüchterntoleranzzeiten, Carnitingaben, Ernährung, Maltodextrin und überhaupt die praktizierte Vorgehensweise zur endgültigen Abklärung des zunächst nur als Verdacht angenommenen MCAD-Mangels.
Einigkeit gab es unter fast allen betreuenden Stoffwechselärzten nur in einem Punkt: wenn man ihnen mitteilte, dass man im Austausch mit anderen betroffenen Familien davon gehört habe, dass an der Uniklinik in der Stadt xy die Empfehlungen zur Ernährung oder die Nüchterntoleranzzeit im aktuellen Alter des eigenen Kindes etwas anders seien, bekam man ein deutlich mitleidiges, um nicht zu sagen gequältes Lächeln geschenkt, zusammen mit der Erwiderung: “Na ja, was soll ich dazu sagen! Seien Sie mal froh, dass Sie hier bei uns betreut werden, denn WIR kennen uns wirklich mit dem MCAD-Mangel aus. Das ist ja offensichtlich längst nicht überall der Fall! Soll ich Ihnen mal sagen, wie oft die dort betreuten Kinder in schwere Stoffwechselkrisen geraten, weil die genannten Nüchternzeiten für das Alter einfach viel zu lang sind / weil man dort kein Carnitin verordnet?”
Ich selbst habe es genau so gesagt bekommen und auch mehrere andere Teilnehmer haben von ähnlichen Reaktionen ihrer Ärzte berichtet. Seltsam war dann nur, dass die Forumsteilnehmer, die an den betreffenden Kliniken betreut wurden, nie von irgendwelchen Entgleisungen berichtet haben. Ich kann mir auch nicht vorstellen, dass eine Uniklinik auch immer sofort einen Info-Rundbrief an alle anderen Stoffwechselzentren schickt: “Hallo zusammen, es ist wieder eines der an unsere SWA betreuten MCAD-Kinder entgleist. Das nur zur Info für euch, damit ihr euren Patienten mal wieder mitteilen könnt, dass die von uns genannten Nüchternzeiten doch zu lang sind, und dass unser Verzicht auf Carnitinverordnungen ebenfalls falsch ist.“
Es anders zu machen als alle anderen, war schon 2008 (und sicher auch schon davor) das Alleinstellungsmerkmal, mit dem man sich an fast allen Unikliniken zu brüsten versuchte. Man darf allerdings nicht dem Irrtum unterliegen, daraus abzuleiten, dass man sich an diesen Unikliniken besonders intensiv mit der Erforschung des MCAD-Mangels beschäftigt und darauf seine individuelle Vorgehensweise aufgebaut hat. Vielmehr basieren alle diese unterschiedlichen Empfehlungen auf schon relativ alten Veröffentlichungen diverser, meist ausländischer Studien aus den Niederlanden oder den USA, in denen z.B. mit überwachten Fastentests ausprobiert worden war, wie lange MCAD-Kinder in unterschiedlichem Alter hungern konnten, bevor bei ihnen klinische Symptome zu bemerken waren. Da man dazu ja überhaupt erstmal Familien finden musste, die sich auf so ein gewagtes Experiment an ihrem eigenen Kind im Dienste der Wissenschaft einlassen wollten, basierten die erhobenen Daten meistens auf ziemlich kleinen Versuchsgruppen mit teilweise nur drei bis fünf Kindern pro Altersgruppe. In manchen Studien wurde zusätzlich berücksichtigt, welche MCAD-Ausprägung aufgrund der gefundenen Genmutationen bei den Kindern vorlagen, bei anderen Versuchen wurde nicht zwischen schweren und milden Varianten unterschieden, sodass es auch Kinder gab, bei denen der Versuch nach über 30 Stunden ohne jegliche Symptome abgebrochen wurde, während andere gleichaltrige Kinder schon nach vielleicht 14 oder 16 Stunden deutliche klinische Symptome zeigten. Diese gezielt durchgeführten Versuche wurden ergänzt durch die zurückliegenden Nüchternzeiten einzelner Kinder, die aufgrund einer Entgleisung in verschiedenen Kliniken behandelt worden waren. Da es sich in solchen Fällen üblicherweise um Krankheitssituationen mit Erbrechen gehandelt hatte, basierten die Angaben zur zurückliegenden Nüchternzeit dann vor allem auf groben Annahmen, da sich nur schwer einschätzen ließ, ob nun wirklich die letzte Mahlzeit als Anfangszeitpunkt gewertet werden konnte oder ob man aufgrund des Erbrechens eher noch eine weitere Mahlzeit zurückdenken musste. Jede dieser Veröffentlichungen enthielt dann auch eine sich aus diesen Beobachtungen ableitende Tabelle mit empfohlenen Nüchternzeiten, bei denen in einem Fall die Obergrenze für ein bestimmtes Alter dann halt mal bei 8 Stunden, in einem anderen Fall aber vielleicht schon bei 10 oder sogar 12 Stunden lag.
Wie gesagt, wurden diese ganzen Studien zur aktiven Ermittlung von sicheren Nüchternzeiten schon vor ziemlich langer Zeit gemacht. Zumindest seit 2008 habe ich von keiner der im Forum vertretenen rund 550 MCAD-Familien von entsprechenden Anfragen gehört. Neuere Studien zu Nüchternzeiten beschränken sich daher vermutlich darauf, die verschiedenen “alten” Daten aus aller Welt zusammenzutragen und anhand dieser somit größeren Datenbasis und eventuellen jüngeren Fällen von eingetretenen Entgleisungen zu überprüfen, ob man die Zeiten-Tabellen eventuell anpassen sollte.
Welche Nüchternzeiten-Übersicht von einer Uniklinik als eigene Empfehlung an die betreuten Familien weitergegeben wurde (und auch heute immer noch wird), hing einfach nur davon ab, auf welche dieser verschiedenen alten Studien und darin erwähnten Tabellen man zuerst gestoßen ist, bzw. welche der verschiedenen Tabellen man am sinnvollsten fand. Hier spielte somit keine übergeordnete wissenschaftliche Erkenntnis, sondern einzig und alleine der Faktor Mensch eine Rolle. Hinzu kommt, dass der MCAD-Mangel aufgrund der nur wenigen pro Jahr hinzukommenden betroffenen Kinder in den meisten Unikliniken nur ein ganz kleines Randthema ist und in den Stoffwechselambulanzen nur mitbehandelt wird, weil er nun mal ins Themengebiet fällt. Man kann sich aber vorstellen, dass gegenüber den an den SWAs betreuten unzähligen an behandlungsbedürftigem Übergewicht oder sogar Adipositas leidenden Kindern, sowie den pro Jahr etwa 3700 neu hinzukommenden Kindern mit Typ-1-Diabetes, die im Vergleich dazu “nur” rund 70 neuen MCAD-Fälle (die sich dann auch noch auf fast drei Dutzend SWAs in ganz Deutschland verteilen) kaum ins Gewicht fallen. Zumindest nicht, wenn es darum geht, die seit Jahrzehnten praktizierten Vorgehensweisen mal komplett zu überdenken und auf aktuellere Erkenntnisse der letzten paar Jahre zu stützen.
Eine gewisse Vereinheitlichung der Empfehlungen gab es mit dem Erscheinen der ersten MCAD-Broschüre von Milupa, die an die Unikliniken geschickt und von einigen Stoffwechselärzten auch an neue MCAD-Familien verteilt wurde. Längst nicht alle Ärzte gaben dieses Heftchen weiter, wie wir im Forum über die Jahre hinweg feststellen konnten. Manchmal wurde sie einem auch nur mit gleichzeitigen mündlichen Sicherheitshinweisen in die Hand gedrückt. Uns wurde z.B. gesagt, dass wir auf die enthaltene Nüchternzeitentabelle nichts geben sollten. Man hätte hier eine eigene, die noch etwas restriktiver und deshalb viel sicherer sei. Auf die tatsächlich bestehenden Nachteile der enthaltenen Tabelle und die zum Teil auch dadurch aufgetretenen Missverständnisse (sie endete z.B. mit einem Alter von 7 Jahren), wird an anderer Stelle noch genauer eingegangen. Keine Probleme mit der Übernahme der Empfehlungen aus dem Heftchen hatte man dagegen an all den SWAs, deren Stoffwechselärzte im Gespräch mit den Eltern immer erst darin nachschlagen mussten, um die ihnen gestellten Fragen beantworten zu können – falls überhaupt etwas dazu in der Broschüre stand. Soviel dazu, dass jede Uniklinik für sich in Anspruch nehmen möchte, dass größte und genaugenommen auch einzige Kompetenzzentrum zum MCAD-Mangel zu sein.
In gleicher Weise verhält es sich mit dem Carnitin. Auch da haben in den vergangenen Jahrzehnten nur wenige SWAs ihre Haltung bzw. Vorgehensweise überdacht und geändert. Manche Ärzte vertreten nach wie vor die Haltung “Viel hilft viel – also rein damit, und außerdem haben wir das hier schon immer so gemacht!” Andere haben immerhin ein Einsehen, dass der oftmals daraus resultierende penetrante Fischgeruch den Kindern im täglichen Umgang mit ihren Mitmenschen erheblich schadet und verschreiben kleinere Portionen oder nur im Einzelfall nach regelmäßigen Kontrollen.
Hierzu ein kleiner Tipp: Wenn du bei der nächsten Kontrolluntersuchung in der SWA erreichen möchtest, dass der überprüfte Carnitinspiegel auf einem möglichst hohen Niveau ist, dann gib deinem Kind die tägliche Dosis etwa 3-4 Stunden vor dem Termin bzw. vor dem zu erwartenden Zeitpunkt der Blutabnahme. Zu diesem Zeitpunkt ist die im Blut feststellbare Menge an freiem Carnitin am höchsten, während sie in den folgend 6-8 Stunden langsam auf ihr normales Basis-Niveau zurückkehrt. Willst du dagegen wissen, wie viel die tägliche Carnitingabe seit der letzten Untersuchung wirklich gebracht hat, dann gib deinem Kind die tägliche Dosis einfach mal erst nach dem Termin in der SWA. Dann ist in der Blutprobe das freie Carnitin auf seinem tatsächlichen Basisniveau feststellbar und nicht der durch die vorherige Einnahme ausgelöste hohe Peak-Wert. Es könnte dann allerdings dazu kommen, dass der aktuelle Carnitinspiegel gegenüber dem letzten Ergebnis von vor einem halben Jahr scheinbar deutlich abgesunken ist und euer Arzt euch deshalb eine höhere tägliche Dosis verordnen will. In Wirklichkeit war aber der in der zurückliegenden Untersuchung gemessene Carnitinwert durch die zuvor erfolgte Einnahme vermutlich auch nur stark in die Höhe gepusht worden und der aktuelle Wert zeigt das wahre Niveau an, wie es durch die fortwährenden Carnitingaben erreicht wurde.
In den vergangenen Jahren haben erfreulicherweise immer mehr SWAs eingesehen, dass die alleine auf der molekulargenetischen Untersuchung beruhende Diagnose des MCAD-Mangels in einigen Fällen doch zu keinem zufriedenstellenden Ergebnis führt. In den meisten Fällen wurde und wird dabei zwar die bekannte häufigste Variante K329E homozygot oder eine andere bereits als schwer (engl. “severe”) bekannte Mutationskombination gefunden, aber dennoch gibt es Jahr für Jahr ein paar Kinder, bei den man aus den gefundenen Mutationen keinen endgültigen Rückschluss auf die zu erwartende Schwere bzw. Milde des MCAD-Mangels ziehen kann. Die früheren Zeiten, in denen sich die Stoffwechselärzte in so einem Fall dann noch vor die Eltern stellten und sinngemäß sagten: “Ist mir doch egal, was ihr Kind hat! Es war im Screening irgendwie auffällig und jetzt wurden ja auch zwei Mutationen gefunden. Also haben Sie als Eltern bitte soviel Sorgen wie bei einem eindeutig schweren MCAD-Mangel und genauso wird ihr Kind für den Rest seines Lebens behandelt.” sollten dank der inzwischen sehr ausgereiften und sehr verlässliche Ergebnisse liefernden Residualaktivitätsanalyse endgültig vorbei sein.
Tatsächlich empfehlen inzwischen immer mehr Unikliniken als nächsten Schritt direkt nach einem immer noch auffälligen Konfirmations-Screening diese Analyse der tatsächlich verbleibenden Leistungsfähigkeit der MCAD-Enzyme – selbst Einrichtungen, die bis vor ein paar Jahren diese Untersuchungsmethode noch rigoros abgelehnt hatten. Manchmal müssen vielleicht auch erst ein paar zuständige Ärzte wechseln, bevor neuere und bessere Methoden etabliert werden können.
So hat sich durch die Residualaktivitätsanalyse schon oft gezeigt, dass Neugeborene mit einem zunächst scheinbar deutlich auf einen MCAD-Mangel hinweisenden Screening-Ergebnis (bei dem von ärztlicher Seite dann meistens gleich ein schwerer MCAD-Mangel in Aussicht gestellt wird), aufgrund der ermittelten sehr hohen Restaktivität von annähernd 50% dann doch nur Carrier sind, oder bei einer im mittleren Bereich liegenden Prozentzahl immerhin einen nur milden MCAD-Mangel haben, bei dem das Risiko für eine Stoffwechselentgleisung gegenüber der bekannten klassischen Variante extrem gering ist. Durch diese Untersuchungsmethode können heutzutage Kinder auch noch ein paar Wochen nach der Geburt sicher vom Verdacht des MCAD-Mangels befreit werden, während früher für viele Stoffwechselärzte bereits nach dem zweiten auffälligen Screening und spätestens nach dem Nachweis von zwei Mutationen – selbst wenn deren Signifikanz unklar war – der Würfel gefallen und der Drops gelutscht war.![]()
Hinsichtlich dieser Frage, ob es bzgl. des MCAD-Mangels extra sinnvolle oder vielleicht auch problematische Lebensmittel gibt, gehen die Meinungen unter den deutschen Stoffwechselärzten mal wieder weit auseinander, und letztlich bleibt es jeder von einem MCAD-Mangel betroffenen Familie selbst überlassen, welche dieser Ansichten man am besten nachvollziehen kann und welcher man sich letztlich anschließen will. Man kann sich aber mit ein wenig Nachdenken auch eine eigene Meinung zu dem Thema bilden, denn es ist nicht besonders schwer, die Zusammenhänge zu verstehen und daraus dann ein paar die Ernährung betreffende Schlüsse zu ziehen.
Die einfachste, von einigen Stoffwechselärzten geäußerte Ansicht, muss man nicht weiter ausführen, denn an “Ihr Kind kann alles essen, Sie müssen überhaupt nichts beachten!” gibt es nicht viel herumzudeuten. Worüber man lediglich diskutieren könnte, ist die Frage, ob diese Aussage aus einer bereits erfolgten tiefergehenden Beschäftigung mit den beim MCAD-Mangel gestörten Stoffwechselvorgängen resultierte oder ob es sich um eine reine Vermutung aufgrund nur geringfügiger MCAD-Kenntnisse handelt.
Um sich dem Thema zu nähern, muss man sich noch einmal ganz grob vor Augen halten, was beim MCAD-Mangel nicht richtig funktioniert und welche Seiteneffekte dies haben kann.
Ganz extrem vereinfacht dargestellt bewirkt der MCAD-Mangel, dass die Verwertung von Fettsäuren im Körper eines davon betroffenen Menschen nach den ersten paar Verarbeitungsschritten bei mittelkettiger Restlänge endet, weil das für die weitere Aufspaltung der Kohlenstoffketten benötigte MCAD-Enzym einen “Mangel” aufweist.
Die meisten in der natürlichen Nahrung vorkommenden Fette bestehen aus langkettigen Fettsäuren, von denen durch die vor dem MCAD-Enzym an die Reihe kommenden Enzyme noch einige Stücke abgespalten und zur Energiegewinnung genutzt werden können. Nur ganz wenige Lebensmittel enthalten neben den langkettigen auch einen Anteil von vorneherein nur mittelkettiger Fettsäuren.
Jede Fettsäurenkette wird aufgrund des MCAD-Mangels im Laufe ihrer Verwertung irgendwann bei mittelkettiger Restlänge enden, mit Hilfe von Carnitin aus den Mitochondrien als Acylcarnitin ausgeschleust und schließlich über die Nieren ausgeschieden werden. Das zum Abtransport aus den Mitochondrien angekoppelte Carnitin geht dabei verloren, denn es verlässt den Körper und steht damit nicht zur weiteren Verwendung zur Verfügung. Diesbezüglich macht es keinen Unterschied, ob ein Lebensmittel ausschließlich langkettige Fettsäuren enthält, oder ob es auch einen gewissen Anteil mittelkettiger Fettsäuren aufweist. Jede einzelne in den Mitochondrien verwertete Fettsäure, egal ob aus der gerade aufgenommenen Nahrung oder aus dem gespeicherten Körperfett, wird als mittelkettiger Rest mitsamt eines Carnitinmoleküls ausgeschieden.
Der Unterschied besteht einfach darin, dass bei der Verarbeitung von langkettigen Fettsäuren ein gewisser Restnutzen durch die zunächst noch abgespaltenen 2er Kohlenstoffketten besteht, während eine zur Verwertung in die Mitochondrien transportierte mittelkettige Fettsäure nur zu einem Carnitinverlust führt, ohne zuvor noch etwas zur Energiegewinnung beizusteuern. In diesem Zusammenhang spielt nur das direkt verwertete Nahrungsfett der täglichen Mahlzeiten eine Rolle, denn das nicht benötigte und per Lipogenese in Körperfett umgewandelte Nahrungsfett wird grundsätzlich in Form von langkettigen Fettsäuren gespeichert. So kann kein mittelkettiges Fett mehr aus den Fettspeichern in die Mitochondrien gelangen.
Soweit die zu berücksichtigenden Hintergründe. Unter diesem Gesichtspunkt erscheint es logisch, dass zur Vermeidung von unnützem Carnitinverlust der Anteil der mittelkettigen Fettsäuren in der täglichen Ernährung möglichst gering gehalten werden sollte. Dies erfordert allerdings keinen großen Aufwand, wenn man ein paar simple Informationen im Hinterkopf behält:
1. von allen natürlichen Produkten enthalten lediglich Kokosöl und Palmkernöl einen großen Anteil mittelkettiger Fettsäuren. In Kokosfett ist der Anteil dagegen deutlich geringer, aber immer noch bei ungefähr 10% der gesamten Fettsäurenmenge. Im Gegensatz zum Palmkernöl wird das heutzutage in sehr vielen Lebensmitteln enthaltene Palmöl/Palmfett, trotz des ähnlich klingenden Namens, nicht aus den Kernen, sondern aus dem Fruchtfleisch der Früchte der Ölpalme gewonnen und besteht fast ausschließlich aus langkettigen Fettsäuren.
2. Das ausschließlich aus mittelkettigen Fettsäuren bestehende MCT-Fett, bzw. MCT-Öl sollte vollständig vermieden werden. Dieses spezielle Produkt wird industriell aus dem bereits erwähnten Kokosöl oder Palmkernöl extrahiert. Man findet es in speziellen Babynahrungsprodukten, die vor allem bei Durchfallproblemen zur Anwendung kommen. Außerdem wird es als MCT-Öl in Reformhäusern verkauft und stellt für Menschen mit VLCAD- oder LCHAD-Mangel einen wichtigen Nahrungsbestandteil dar, da diese keine langkettigen Fettsäuren verarbeiten können und entsprechende Fette in der Nahrung nach Möglichkeit vermeiden müssen. Bezüglich des Vorkommens in der Nahrung kommt regelmäßig die Frage auf, ob die normale verwendete Babynahrung nicht unbemerkt ebenfalls MCT-Öl enthalten könne, wenn z.B. mit einer besonders guten Verdaubarkeit geworben wird. Die Verwendung von MCT-Öl wird aber seitens aller Hersteller als besonderes Merkmal herausgestellt und deshalb auf den dieses Fett enthaltenen Produkten mit besonders auffälliger Erwähnung auf der Verpackung hervorgehoben. Sofern eine Verpackung also nicht ausdrücklich und ganz deutlich erwähnt, dass MCT enthalten ist, braucht man sich keine Gedanken darum machen, ob es vielleicht doch drin sein könnte. Im Zweifelsfall fragt man aber einfach vor der Auswahl eines Produktes in der Apotheke nach dessen Inhaltsstoffen und ob MCT oder “mittelkettige Triglyceride” enthalten sind.
Abschließend zu diesem Punkt muss aber eines noch ganz deutlich herausgestellt werden: Die vereinzelte Aufnahme geringer Mengen mittelkettiger Fette stellt für Kinder mit MCAD-Mangel keine Gefahr dar, und man muss keine Angst davor haben, seinem Kind durch ein paar vielleicht versehentlich oder unbewusst in der Nahrung enthaltene mittelkettige Fette schwer zu schaden. Weder Kokosöl, Kokosfett, Palmkernöl noch sogar reines MCT-Öl sind unmittelbar schädlich, wenn diese gelegentlich und in geringfügigen Mengen mit der Nahrung aufgenommen werden. Sie werden auch keine Entgleisung begünstigen oder gar hervorrufen. Wie oben erwähnt, werden im Verlauf der Fettsäurenverwertung grundsätzlich alle Fette irgendwann zu mittelkettigen Fettsäuren verkürzt. Im Hinterkopf behalten sollte man nur den Umstand, dass die von vorneherein nur mittelkettig vorliegenden Fettsäuren selbst keinen Restnutzen zur Energiebereitstellung leisten können, sondern zwecks Ausscheidung lediglich Carnitin verbrauchen, was nach Möglichkeit vermieden werden sollte.![]() Weitere Informationen findest du im Artikel “Ernährung beim MCAD-Mangel“.
Weitere Informationen findest du im Artikel “Ernährung beim MCAD-Mangel“.
Um schon mal die vielleicht aufkommende Verwirrung zu beseitigen: Falls dein Kind in den Jahren nach 2014 geboren wurde, ist dir die hier und in den weiteren Artikeln öfters angesprochene Milupa-Broschüre vermutlich eher als MCAD-Broschüre von Nutricia Metabolics bekannt, was alleine daran liegt, dass sich die Firma Milupa im Jahr 2015 in Milupa Nutricia umbenannt hat und unter diesem Label dann auch eine neue Version der MCAD-Broschüre aufgelegt hat. Zumindest früher wurde den Eltern die damals noch unter dem Label Milupa Metabolics herausgegebene Broschüre beim ersten Besuch in ihrer Stoffwechselambulanz, mit den Worten “Da steht alles drin, was sie wissen müssen!” in die Hand gedrückt. Leider schienen sich auch die MCAD-Kenntnisse einiger dieser Ärzte ziemlich genau auf den Inhalt dieses kleinen Heftchens zu beschränken.
Da ich die meisten Artikel dieser Seite in den Jahren 2008 bis 2015 geschrieben habe, sind die hier und da zu findenden Verweise auf die Inhalte dieser “alten” Broschüre von Milupa bezogen.
In der aktuellen Version wurde das Info-Heftchen von sich wirklich hervorragend auskennenden Stoffwechsel-Ärztinnen der Freiburger Kinderklinik vollständig überarbeitet, neu strukturiert und in einigen Punkten präzisiert, sodass man als ganz neu betroffene Eltern einen wirklich guten ersten Einstieg in das Thema erhalten kann.
Vor allem aber gefällt mir sehr gut, dass auf der letzten Seite nun endlich ein auf festerem Papier gedruckter, richtig ordentlich und seriös aussehender MCAD-Notfallausweis in den Sprachen Deutsch und Englisch zum Heraustrennen enthalten ist. In diesen müssen dann nur noch die Daten des Kindes eingetragen werden und dann sollte man ihn nach Möglichkeit immer mit sich führen. Ein Notfallausweis war zwar auch schon in der alten Milupa-Broschüre vorhanden, allerdings wurde dieser, den damaligen Schilderungen einiger Eltern zufolge, in der Notaufnahme einiger Krankenhäuser keines zweiten Blickes gewürdigt und von manchem Arzt so ernst genommen, als hätte man ihm den Detektivausweis aus einem Micky-Maus-Heft vorgelegt − was in Wahrheit aber weniger einen Rückschluss auf die Qualität des alten Ausweises, als auf die der ihn ignorierenden Ärzte zulässt.
Falls du das aktuelle Heftchen nicht bereits in deiner Stoffwechselambulanz ausgehändigt bekommen hast, kannst du es über die Webseite von Nutricia Metabolics kostenlos bestellen. Dies ist allerdings nicht mehr ganz so einfach wie zu früheren Zeiten! Im Service-Bereich ist unter dem Punkt “Informationen für den Alltag” ein Login-Button für den Zugang zum Communitybereich zu finden. Hier musst du dich dann leider auch schon mit ein paar Daten und einer existierenden E-Mail-Adresse registrieren. An diese Adresse erhältst du dann kurz darauf eine Verifikations-E-Mail, in der du einen Bestätigungslink anklicken musst, damit dein Community-Profil freigeschaltet wird. Sobald Du dann eingeloggt bist, erscheint unter den Alltagsinformationen eine aufklappbare Rubrik “Andere seltene Stoffwechselstörungen”, unter der Du dann endlich auch die MCAD-Broschüre und einen Anforderungs-Link findest. Eine Downloadmöglichkeit gibt es bedauerlicherweise nicht. Die Broschüre kann nur postalisch bestellt werden, sodass du zwingend auch noch deinen echten Namen und deine komplette Anschrift hinterlassen musst. Zusätzlich musst du noch angeben, inwiefern Du mit dem MCAD-Mangel zu tun hast (Patient, Elternteil, Verwandter, Ernährungsfachkraft, Arzt). Wenn man − so wie ich − dort “Andere” auswählt, wird seitens Nutricia zunächst telefonisch oder per E-Mail noch mal genau nachgefragt, weshalb man sich für die MCAD-Broschüre interessiert, damit man optimal beraten werden kann. Natürlich auch, damit sich keine völlig unbeteiligten Personen die ganzen Heftchen aus purem Interesse abgreifen, sondern sie den wirklich davon betroffenen Familien vorbehalten bleiben. Ich vermute aber, dass man in jedem Fall von Nutricia kontaktiert würde.
Meiner persönlichen Meinung nach ist es durchaus sinnvoll, die Broschüre anzufordern, um die wichtigsten Informationen zum MCAD-Mangel in einer so kompakten Form schriftlich vorliegen zu haben, dass man sie vielleicht auch mal den Großeltern oder Freunden erläutern kann. Sollte es Dir aber bzgl. des Bestellvorganges der Broschüre an irgendeiner Stelle mit der Preisgabe persönlicher Daten zu viel werden, kann ich Dir versichern, dass du in dem Heftchen keine über die Inhalte dieser Webseite hinausgehenden Informationen finden wirst. Letzten Endes ist es, so wie auch die frühere Milupa-Broschüre, ein Info-Heftchen für absolute MCAD-Neulinge. In dieser Rolle ist es aber aus meiner Sicht absolut empfehlenswert − nicht zuletzt wegen des wirklich gut gestalteten Notfallausweises.![]()
Wer die Situation nicht selbst schon hinter sich hat, wird sie in nicht allzu ferner Zukunft mit Sicherheit selbst erleben. Spätestens wenn dein Kind in den Kindergarten kommt, siehst du dich der Herausforderung ausgesetzt, den Erzieherinnen in relativ kurzer Zeit und doch ausführlich genug eine Erklärung bieten zu müssen, was sich hinter diesem ihnen höchstwahrscheinlich völlig unbekannten MCAD-Mangel verbirgt, an dem dein Kind vermeintlich “leidet”.
Selbst wenn du es schaffst, ihnen die bei deinem Kind gestörten Stoffwechselvorgänge und daraus resultierenden möglichen Risiken auf sehr niedrigem Niveau zu erläutern (dazu gibt es im Downloadbereich einen auf dein Kind anpassbaren Infobrief), wird bei den meisten Menschen doch nur eine Erkenntnis hängen bleiben: das Kind ist irgendwie krank.
Dies ist auch sehr verständlich, denn wenn man sich an an die ersten Tage und Wochen nach der Befundmitteilung über den MCAD-Mangel des eigenen Kindes zurückerinnert, wird man feststellen, dass man nach der Erstinformation zunächst mal auch das Verständnis hatte, dass das Kind krank, vielleicht sogar schwer krank sei. Dass es aber “nur” eine Stoffwechselstörung hat, und dies schon alleine aus medizinischer Sicht etwas völlig anderes ist, als eine chronische Krankheit, ist eine Sichtweise, zu der man erst einmal selbst gelangen musste. Dieser Schwenk von “mein Kind ist krank” zu “mein Kind hat zwar eine Stoffwechselstörung, aber ist nicht chronisch krank” erforderte eine gewisse Reifung, die ein paar Wochen, oder sogar Monate dauerte. So etwas werden die Erzieherinnen der Kita jedoch nicht erleben, denn sie haben weder die Zeit, noch die Motivation, sich so intensiv mit den medizinischen Hintergründen des − äh, wie hieß die Krankheit nochmal − zu befassen, dass sich bei Ihnen von selbst dieser Wechsel der Sichtweise einstellen könnte.
Nun, genaugenommen kann es dir eigentlich egal sein, ob die Erzieher im Kindergarten, oder später die Lehrer in der Schule die wahre Natur des MCAD-Mangels verstehen, oder nicht, solange sie nur wissen, wie sie im Notfall zu reagieren haben, und ansonsten dein Kind ganz normal, also wie jedes andere auch behandeln.
Es haben aber Eltern auch schon die Erfahrung gemacht, dass ihr Kind vom ersten Moment bei den Erzieherinnen den “dieses Kind ist krank”-Stempel aufgedrückt bekam, weil dies nun mal das Verständnis war, das sich aus dem Vorgespräch bei ihnen eingebrannt hatte. Ein Kind mit MCAD-Mangel ist aber im medizinischen Sinne nicht in einem dauerhaft kranken Zustand, daher sollte ihm auch nicht ungerechtfertigter Weise von Dritten dieser Stempel aufgedrückt werden. Schon gar nicht sollte dieses falsche Verständnis dazu führen, dass das Kind innerhalb der Kita-Gruppe oder der Schulklasse als “das kranke Kind” herausgestellt wird, aufgrund dessen ihm eine Sonderbehandlung gewährt werden muss. Das hat es nun wirklich nicht verdient, und das ist auch nicht gerechtfertigt.
Das Vorhandensein einer Stoffwechselstörung bedeutet, dass im Körper eines davon betroffenen Menschen etwas nicht ganz so, wie bei den meisten anderen Menschen funktioniert. Das trifft aber z.B. auch auf einen blinden Menschen zu, dessen Sehsinn − aus welchem medizinischen Grund auch immer − nicht funktioniert. Dies bezeichnet man dann zwar als Behinderung, aber nicht als Kranksein, und auch kein blinder Mensch würde von sich selbst sagen, dass er sich durch die Blindheit krank fühle. Natürlich muss er mit den ihm verbleibenden Sinnen vermehrt auf seine Umgebung achten und Vorsichtsmaßnahmen treffen, um sich nicht zu verletzen oder durch einen Unfall zu schaden zu kommen, aber alleine der Umstand blind zu sein, bedeutet nicht, dass man auch krank sei.
So bedeutet auch das Vorliegen des MCAD-Mangels im Körper eines davon betroffenen Kindes für sich selbst genommen nicht, dass dieses Kind deshalb krank ist. Etwas verwirrend ist in diesem ganzen Zusammenhang allerdings schon, dass der in engem Zusammenhang stehende Begriff “Krankheit” definiert wird als “Störung der Funktion eines Organs, der Psyche oder des gesamten Organismus.” Nach dieser Definition ist jegliche Abweichung von der optimalen Gesundheit eine Krankheit, und auch der zuvor erwähnte Blinde würde insofern an einer Krankheit leiden, auch wenn er sich selbst nicht als krank empfindet.
In diesem rein medizinischen Sinne kann der MCAD-Mangel also durchaus als Krankheit im Sinne der Störung einer gewissen organischen Funktion bezeichnet werden. Dies bedeutet allerdings nicht, dass ein davon betroffener Mensch als chronisch und somit ständig krank zu bezeichnen wäre. Normalerweise ist nämlich selbst ein Kind mit MCAD-Mangel so gesund, wie jedes andere Kind auch, kann genau das gleiche leisten und genauso herumtoben, wie alle seine im Vergleich zu ihm vermeintlich “gesunden” Altersgenossen. Vor allem aber ist es nicht “ansteckend”! Auch das ist etwas, was vor allem kleine Kinder mit dem Kranksein in Verbindung bringen. Wenn sie krank sind, müssen sie zu Hause bleiben, damit sie andere Kinder in Kita oder Schule nicht anstecken − sonst werden die auch krank. Oder noch schlimmer − sie selbst sind jetzt krank geworden, weil ein anderes Kind in ihrer Gruppe krank ist oder war, und sie sich bei ihm angesteckt haben. Ein Kind, was von den Erziehern gegenüber der Gruppe pauschal als krank dargestellt wird, ist immer irgendwie anders. Vor allem dann, wenn es jeden Tag auch zwischendurch mal ein Brötchen oder einen Müsliriegel essen darf, was den anderen Kindern der Gruppe vielleicht nicht zugestanden wird. Wenn diese dann nachfragen, warum der und nicht sie, und sie dann zur Antwort bekommen: “Der X. ist krank, der darf das!” wird diese ungerechtfertigte und falsche Herausstellung der kleinen Besonderheit des Kindes ständig wieder neu aufgefrischt.
Dein Kind ist nicht krank, sondern normalerweise absolut gesund. Es kann wie jedes andere Kind durch Krankheitserreger krank werden, und aufgrund des MCAD-Mangels kann es unter gewissen Gegebenheiten auch sehr viel kränker werden, als andere Kinder, aber dies ist eine ganz seltene Ausnahme und nicht der Normalzustand.
Wie kann man diese viel mehr den Tatsachen entsprechende Sicht der Dinge aber anderen Personen vermitteln, die über so gut wie kein Hintergrundwissen verfügen, und auch längst nicht die Zeit haben, um durch eigene Überlegungen zu dieser gänzlich anderen Sichtweise zu gelangen?
Ist der MCAD-Mangel eine Allergie? Nein, er ist gewissermaßen genau das Gegenteil, aber wenn man Menschen ohne jegliche Vorkenntnis den MCAD-Mangel erklären will, ist es sinnvoll, sie anhand etwas allgemein Bekanntem gedanklich abzuholen und von dort aus den dann sehr viel leichter nachvollziehbaren Schwenk zu den Besonderheiten des MCAD-Mangels durchzuführen.
Von Allergien sind sehr viele Menschen betroffen. Es ist ein sehr häufig und sehr ausführlich in den Medien behandeltes Thema, mit dem die Allgemeinheit somit bestens vertraut ist. Daher dürfte niemand Probleme damit haben, die folgende Eklärung nachzuvollziehen, mit der Du die kleine Besonderheit deines Kindes in einem Gespräch mit den Erzieherinnen verdeutlichen könntest:
Hat jemand eine Allergie auf z.B. Erdnüsse, dann wird er krank (im hier verwendeten umgangssprachlichen Sinn), wenn er Erdnüsse zu sich nimmt. Es kann für ihn dann aufgrund der Erdnüsse sogar richtig gefährlich werden. Solange er aber ganz gezielt und bewusst auf das Essen von Erdnüssen verzichtet, ist er nicht im geringsten krank, sondern völlig gesund − so gesund, wie jeder andere Mensch auch. So verhält es sich bei jeder Form von Allergie oder Unverträglichkeit. Kommt der Allergiker mit dem für ihn allergieauslösenden Stoff in Berührung, wird er krank. Solange er diesen Stoff konsequent meidet, ist er − zumindest in dieser Hinsicht − so gesund, wie man nur sein kann.
Bei deinem Kind ist es nun genau entgegengesetzt. Eine Stoffwechselstörung wie der MCAD-Mangel ist gewissermaßen genau die Umkehrung einer Allergie. Dein Kind ist normalerweise völlig gesund − so gesund wie alle anderen Kinder in der Kita auch. Damit das aber auch ganz sicher so bleibt, muss es (im Gegensatz zum Nahrungsallergiker, der auf gewisse Lebensmittel gezielt verzichten muss, und trotzdem nicht “krank” ist) aber regelmäßig bestimmte Sachen essen, dazu gehören z.B. Brötchen, Müsliriegel, Obst, gesüßte Getränke usw. Nur dann, wenn es das nicht macht, kann es unter gewissen Umständen (z.B. stark anstrengende Aktivitäten und dabei ausbleibende Nahrungsaufnahme) passieren, dass ihm die Energie ausgeht, und es infolgedessen “krank” wird.
Du solltest gegenüber den Erzieherinnen auch ausdrücklich erwähnen, dass diese mit dem MCAD-Mangel zusammenhängenden, schon von jeher nur seltenen Krankheitssituationen, heutzutage bei fast keinem davon betroffenen Kind mehr auftreten, da man inzwischen sehr genau weiß, wie einfach und wirksam die Prävention ist. Allergiker müssen bestimmte Nahrungsmittel gezielt vermeiden, Menschen mit MCAD müssen zwischendurch mal was essen.
Dass sich dieses “zwischendurch mal was essen” erfahrungsgemäß in manchen Phasen gar nicht so einfach darstellt, wie es sich anhört, muss man in dem Zusammenhang ja nicht auch noch erwähnen.
In gleicher Weise kann man den MCAD-Mangel auch kleinen Kindern erklären, allerdings sollte dies dann anhand eines oder mehrerer konkreter und bekannter Beispiele erfolgen und nicht anhand eines abstrakten Allergiebegriffs. Im günstigsten Fall gibt es in der Kita-Gruppe deines Kindes noch andere Kinder, die aufgrund einer Allergie oder Nahrungsmittelunverträglichkeit auf bestimmte Lebensmittel verzichten müssen.
Die Erzieherinnen könnten dann beispielsweise im morgendlichen Stuhlkreis das Thema auf folgende Weise ansprechen:
Ihr wisst ja alle, dass Alina keine Milch trinken darf, weil es ihr sonst nicht gut geht, und ihr schlecht werden kann. Ihr anderen könnt Milch trinken, und euch wird davon nicht schlecht. Und Bastian darf nichts essen, wo Eier drinnen sind, sonst geht es ihm auch nicht mehr gut. Und Carolin hat bei Geburtstagen immer Muffins aus Dinkelmehl dabei, weil sie von Brot oder Kuchen mit Weizenmehl nichts essen darf, weil diese Sachen nicht gut für sie sind.
Bei Dustin ist das so ähnlich, aber während Alina, Bastian und Carolin bestimmte Sachen nicht essen dürfen, weil es ihnen sonst nicht mehr gut geht, oder ihnen davon schlecht wird, muss Dustin zwischendurch bestimmte Sachen essen, z.B. ein Brötchen oder auch mal einen Müsliriegel, sonst kann es passieren, dass es ihm auch nicht mehr gut geht.
Das ist schon alles. Mehr müssen die Kinder nicht wissen, und auf diesem Niveau können es selbst Kindergartenkinder problemlos verstehen und auch akzeptieren. Weil es oft auch in der Familie, in der Verwandtschaft, oder auch im Freundes- oder Bekanntenkreis so häufig vorkommt, dass jemand gegen irgendetwas allergisch ist, oder eine Unverträglichkeit gegen bestimmte Nahrungsbestandteile hat, sind die meisten Kinder schon mal irgendwo mit diesem Thema in Berührung gekommen und haben meist keinerlei Problem damit, dass es auch in ihrer Kindergartengruppe einzelne Kinder gibt, die bestimmte Sachen nicht essen dürfen − und die gelten unter ihnen und den Erzieherinnen nicht im geringsten als “krank”.
Auch kleine Kinder erkennen schon den deutlichen Unterschied zwischen den Aussagen “damit es dem Kind auch weiter gut geht, muss es zwischendurch was essen” und “das Kind ist krank und muss deshalb zwischendurch immer was essen”.![]()
Mit ganz wenigen Ausnahmen haben hier alle Eltern die gleiche Erfahrung mit Versicherungen gemacht! Wenn sie irgendwann eine private Krankenzusatzversicherung für ihr Kind abschließen wollen, werden sie von den meisten Versicherungsgesellschaften schlichtweg abgelehnt. In seltenen Fällen wird ihnen die Versicherung zwar angeboten, dann aber nur mit einem hohen Risikoaufschlag, der locker das Doppelte bis Vierfache des normalen Tarifs ausmachen kann. Eltern, die generell privat krankenversichert sind, bekommen manchmal bereits beim Abschluss der normalen Krankenversicherung für ihr Kind hohe Aufschläge aufgedrückt. Die Versicherer sehen in dem Vorliegen einer genetisch bedingten Stoffwechselstörung ein hohes Risikopotential, das sie sich entsprechend bezahlen lassen wollen.
Falls jemand unter diesem Gesichtspunkt mit dem Gedanken spielt, den MCAD-Mangel einfach gegenüber der Versicherung zu verschweigen, ist dies definitiv keine gute Idee! Aufgrund der nun mal im Rahmen der MCAD-Therapie erfolgenden Untersuchungen und Behandlungen, wird die KK ohnehin sehr schnell feststellen, wofür die zur Erstattung eingereichten Kosten angefallen sind und dann u.U. von irgendeinem Sonderkündigungsrecht aufgrund bewusst verschwiegener, relevanter Vorerkrankungen Gebrauch machen und den Versicherungsvertrag ihrerseits kündigen. Dadurch bleiben die Eltern auf ihren Kosten sitzen und müssen sich dann wieder auf die mühsame Suche nach einer neuen Versicherung machen. Dies gestaltet sich dann aber noch sehr viel schwieriger. Mehr dazu weiter unten.
In der gesamten Forenzeit hatte sich bzgl. dieses großen Problems auch keine wirkliche Lösung herausgestellt. Der Abschluss einer privaten (Zusatz-)Versicherung ist schon für die Eltern von Kindern mit MCAD-Mangel meist mit einer langwierigen Suche und vielen Enttäuschungen verbunden − und für die irgendwann sich selbst versichernden MCADler dann erst recht.
Inzwischen habe ich aber einen sehr hilfreichen Tipp von einem diesbezüglich erfahrenen Arzt bekommen – mit der Erlaubnis, diesen hier weiterzugeben.
Es ist seit langem bekannt, dass es auch bei Versicherern eine “schwarze Liste” gibt, das sogenannte Hinweis- und Informationssystem (HIS) des Gesamtverbandes der Deutschen Versicherungswirtschaft (GDV). Vergleichbar mit der Schufa, die Kreditanbietern einen Einblick in die Kreditwürdigkeit eines Antragstellers bietet und bei negativen Einträgen dem Kreditanbieter von einem Vertragsabschluss abrät, gibt es mit dem HIS auch einen entsprechenden, wenn auch in der Öffentlichkeit nicht genauso bekannten Service für Versicherungsanbieter. Wer einmal auf dieser Blacklist gelandet ist, hat so gut wie keine Chance mehr, irgendwo einen neuen Versicherungsvertrag abschließen zu können. Das betrifft vor allem Versicherungskunden, die bereits durch teure Medikamente oder häufige Arztbesuche sehr hohe Kosten verursacht haben und die eine andere private Versicherung deshalb lieber nicht zu ihren Kunden zählen möchte. Anbieter von Berufsunfähigkeits-Versicherungen möchten dagegen keine Kunden mit Rückenleiden oder psychischen Problemen. Selbst die im Kindesalter gestellte Diagnose “übermäßiges Heimweh” kann schon dazu führen, dass einem jungen Menschen zu einem späteren Zeitpunkt die Aufnahme in eine solche Versicherung verwehrt wird.
Man kann aber auch schon auf der Blacklist landen, ohne überhaupt etwas in diesem Sinne “falsch” gemacht zu haben. Es reicht bereits aus, ein einziges Mal von einer Versicherung – aus welchem Grund auch immer – abgelehnt worden zu sein. Der betreffende Antragsteller wird dann quasi als “unerwünschter Kunde” notiert und die Sachbearbeiter anderer Versichungen denken sich infolgedessen: “Wenn er schon von anderen Versicherungen abgelehnt wurde, wollen wir uns den lieber auch nicht ans Bein binden!” . Oftmals hängt es dabei auch nicht an der Sicht des einen Antragsbearbeiters, sondern es gibt versicherungsinterne Regelungen, nach denen auf der Blacklist des HIS geführte Personen nicht aufgenommen werden dürfen. Dann spielt es auch keine wirkliche Rolle mehr, ob die erste Ablehnung vielleicht einfach nur erfolgte, weil der Sachbearbeiter bzw. die Versicherungsgesellschaft den MCAD-Mangel viel schlimmer einschätzte, als es bei anderen Anbietern gesehen würde. Der als Vorerkrankung angegebene MCAD-Mangel ist dann vielleicht sogar von nachrangiger Bedeutung, sondern ausschlaggebend ist der Umstand, dass der Antragsteller zuvor bereits woanders abgelehnt wurde.
Aus diesem Grund gibt es in Versicherungsanträgen fast immer ein Ankreuzfeld “Wurden Sie schon einmal von einer Versicherung abgelehnt?” mit den Auswahlmöglichkeiten Ja und Nein, noch nie abgelehnt. Kreuzt man hier nach einem bereits erlebten Fehlschlag wahrheitsgemäß Ja an, ist der Antrag bereits so gut wie abgelehnt. Kreuzt man dagegen absichtlich Nein an, wird über die generell erfolgende Abfrage beim HIS schnell herausgefunden, ob dies den Tatsachen entspricht – mit entsprechenden Folgen.
Du kannst dir sicher vorstellen, dass die Chance, aufgrund des MCAD-Mangels gleich von der ersten Versicherung abgelehnt zu werden und damit den Weg für sämtliche weitere Versuche bei anderen Gesellschaften verbaut zu bekommen, sehr groß ist. Hat man damit erst einmal diesen Abgelehnt-Makel im HIS an sich, bzw. seinem Kind hängen, ist es zu spät. Aus diesem Grund ist es für dich sehr zu empfehlen, dich an einen Versicherungsmakler, statt direkt an eine Versicherung zu wenden. Der Versicherungsmakler hat für jede gewünschte Versicherungsart (z.B. die Krankenzusatzversicherung) üblicherweise gleich einen ganzen Stapel von Anbietern im Portfolio, bei denen er eine entsprechende Abfrage parallel und vor allem anonymisiert stellen kann. In allen diesen gleichzeitigen Anträgen kann dann wahrheitsgemäß angegeben werden, dass man noch nie von einer Versicherung abgelehnt wurde.
Die Zu- und Absagen kommen dann ebenfalls annähernd gleichzeitig. Sobald eine oder mehrere Versicherungen die Anfrage tatsächlich positiv beantworten, kann man unter diesen Angeboten eventuell noch kurz vergleichen, wie es mit den im Tarif verlangten Zuschlägen aufgrund des MCAD-Mangels aussieht. Dann sollte man aber auch unbedingt zuschlagen. Vor allem, wenn es tatsächlich nur eine Zusage gibt, ist von weiterem Hinauszögern dringend abzuraten. Besser wird es mit weiterem Suchen mit großer Wahrscheinlichkeit doch nicht mehr werden.
Vergleichsportale im Internet sollte man diesbezüglich meiden, denn ohne Angabe der persönlichen Daten geht da für konkrete Angebotseinholungen meistens ohnehin nichts. Gerade auf diese Weise kann man sehr schnell mit seinem Namen auf der Liste der abgelehnten und damit bei allen Versicherungsgesellschaften unerwünschten Kunden landen.
Mittels der zunächst anonymisierten Anfrage durch den Versicherungs-Makler erfahren zwar alle Anbieter von dem zu berücksichtigenden MCAD-Mangel, aber nur die zustimmende und schließlich ausgewählte Versicherung erfährt euch den Namen deines Kindes. Alle ablehnenden Versicherungen kennen ihn nicht und so kann er auch nicht auf die Liste des HIS kommen.
Im Austausch der früheren Forumsmitglieder hat sich gezeigt, dass es bestimmte Situationen, oder − ganz allgemein − bestimmte Aspekte im Leben eines MCAD-Betroffenen gibt, die gewisse Nachteile mit sich bringen, die es ohne das Vorliegen des MCAD-Mangels in dieser Form nicht gäbe.
Diese Nachteile hängen nicht immer speziell mit dem MCAD-Mangel zusammen, denn Menschen mit andern Stoffwechselstörungen oder anderen seltenen Erkrankungen wären meist in gleicher Weise benachteiligt. Aber oftmals spielt es dann doch eine Rolle, dass der im täglichen Leben eigentlich sehr gut zu handhabende MCAD-Mangel den meisten Menschen nun mal unbekannt ist. Die Diabeteserkrankung eines Kindes im Griff zu behalten, bedeutet dagegen Tag für Tag einen fortwährenden und nicht zu unterschätzenden Aufwand. Jedoch ist Diabetes so weit verbreitet und den Menschen, mit denen man zu tun hat (vermeintlich) so gut bekannt, dass diese Stoffwechselstörung für Außenstehende vergleichsweise harmlos erscheint, während dem MCAD-Mangel und seinen potentiellen Auswirkungen (die sich nicht allzu sehr von einem falsch oder unbehandelten Diabetes unterscheiden) irgendwas Dunkles und Bedrohliches anhaftet. Daher gibt es doch gewisse Situationen, in denen die in Kauf zu nehmenden Nachteile für einen MCADler stärker ausfallen, als z.B. für einen Diabetiker.
Die Frage, ob du dich mit andern Eltern austauschen solltest, würde ich jederzeit ganz klar mit “JA” beantworten. Du kannst nur von anderen selbst betroffenen Eltern praxisbezogene Erfahrungen aus dem täglichen Umgang mit einem vom MCAD-Mangel betroffenen Kind bekommen. Also noch nicht mal von mir, denn auch wenn wir selbst für einige Monate gemäß der Aussage unserer Ärzte in dem festen Glauben lebten, unser Sohn habe einen schweren MCAD-Mangel, war das ja letztlich doch nicht der Fall und somit bestand auch zu keinem Zeitpunkt die Gefahr, dass ihm in Krankheitszeiten mit Nahrungsverweigerung etwas hätte passieren können. Er hatte in den gesamten 14 Monaten bis zum endgültigen Abschluss des Nachweises, dass er doch nur Carrier ist, auch keine einzige Krankheitsphase, in der es mit der Ernährung einmal schwierig geworden wäre. Die dir vertrauten Ängste und Sorgen waren zwar auch bei uns ständig im Hinterkopf präsent, aber es kam nie so weit, dass wir selbst vor der schweren Entscheidung standen, ob wir mit ihm zur Stoffwechselklinik zwecks Infusion fahren sollten, oder nicht.
Die Ärzte, mit denen du in der Stoffwechselambulanz zu tun hast oder zu tun bekommen wirst, kennen die ganzen Sorgen aber erst recht nicht, bzw. einzig und alleine aus den Schilderungen der sie um Rat fragenden Eltern und können daher überhaupt keine aus dem täglichen Leben stammenden Erfahrungen weitergeben. Entweder hast du das bereits selbst bemerkt oder wirst es noch feststellen und dir spätestens dann wünschen, dich mit anderen Familien über die konkreten täglichen Situationen austauschen zu können.
Anhand der im Januar 2020 neu aufgenommenen MCAD-Map kannst du mit einem Blick sehen, ob jemand von den anderen Eltern, mit dem Willen zum gegenseitigen Austausch, vielleicht in erreichbarer Nähe zu dir wohnt. In der Vergangenheit hat sich gezeigt, dass der persönliche Austausch mit anderen Eltern eine ganz besondere Qualität hat und weit über jedem rein schriftlichen Austausch steht. Sich einfach mal zum Kaffee oder auf dem Spielplatz zu treffen und ein Stündchen gemütlich miteinander zu plaudern, trägt immens zur Beruhigung bei und man kann über sehr vieles reden, was beim Schreiben einfach viel zu lange dauern würde.
Wie du dich selbst in die MCAD-Map eintragen lässt und Kontakt zu den Familien in den in der Nähe liegenden Orten aufnehmen kannst, ist unterhalb der Karte beschrieben. Natürlich kann es sein, dass man dann letzten Endes doch nicht so ganz kompatibel zueinander ist, aber in der zurückliegenden Zeit des Forums sind auf diese Weise schon bleibende Freundschaften entstanden.
Achtung, ganz entscheidend: diese Aktion lebt vom Mitmachen! Der Seitenzähler im Backend zeigt mir, dass die MCAD-Map tatsächlich sehr häufig aufgerufen wird! Trotzdem wurden fast alle der jetzt dort zu findenden Marker bereits in den ersten zwei Wochen nach der Einrichtung der Karte im Januar 2020 angelegt – größtenteils aus den Reihen ehemaliger Forenmitglieder. Es nützt aber gerade neuen MCAD-Familien nichts, nach vielleicht neuen Markern im eigenen noch leeren Umkreis zu suchen, wenn man selbst keinen eigenen Marker aufnehmen lässt, denn vermutlich warten auch viele andere MCAD-Familien, dass erstmal andere Leute aus ihrer Umgebung den Anfang machen.
Vor ein paar Jahren waren auf der damaligen Foren-Karte über 200 MCAD-Mitglieder markiert, also fünfmal so viele wie jetzt – und selbst das war nur etwa ein Fünftel der in Deutschland lebenden Familien, die zu dem Zeitpunkt vom MCAD-Mangel ihres Kindes wussten. Überall in Deutschland gab es rote Marker, nicht nur in ein paar Ballungszentren. Wieviel diese Karte zur Kontaktaufnahme bringt, hängt also auch heute ganz entscheidend davon ab, dass viele Familien ihre Wohnorte dort eintragen lassen. Erst dann zeigt sich wieder so eindrücklich wie damals, dass man mit der seltenen Stoffwechselstörung seines Kindes nicht alleine ist und dass in gleicher Weise betroffene Familien gar nicht weit weg wohnen. Ein paar neue Kontakte und daraus folgende persönliche Treffen kamen durch die MCAD-Map inzwischen schon zustande und wie ich aus einzelnen Rückmeldungen erfahren habe, sind die betreffenden Familien sehr froh darum. Dieses Angebot darf aber gerne noch viel öfter genutzt werden.
Direkt vorweg: Bei Fragen zu den Inhalten von dieser Seite hier (mcad-infos.de), schreibe mich bitte direkt per Email an (), denn ich bin weder Mitglied der Facebook- noch der WhatsApp-Gruppe. Beides sind aus den Reihen der früheren Forums-Teilnehmer hervorgegangene Initiativen, die in keiner Weise mit mir oder den Inhalten von mcad-infos.de in Verbindung stehen.
In diesem Abschnitt möchte ich gewissermaßen den Beipackzettel mit den Nebenwirkungen beschreiben, damit du im Voraus überlegen kannst, ob du dich selbst schon psychisch in der Lage siehst, mit einer größeren Anzahl anderer betroffener Familien konfrontiert zu werden, denn das ist absolut nicht ohne! Obwohl ein Austausch zwischen MCAD-Betroffenen ohne Frage empfehlenswert und hilfreich ist, macht es dennoch einen großen Unterschied, ob man sich mit ein oder zwei anderen Familien austauscht, oder man es mit 20 oder noch mehr Personen zu tun bekommt. Einen ganz entscheidenden Unterschied macht vor allem, ob du selbst schon einige Zeit mit dem MCAD-Mangel deines Kindes zu tun hast und dabei bereits zu einer gewissen Abgeklärtheit gekommen bist, oder ob dein Denken und Empfinden – wie es gerade bei neu betroffenen Eltern sehr oft der Fall ist – noch weitgehend von Ängsten und Sorgen geprägt ist. In letzterem Fall würde ich dir raten, zunächst einmal den Kontakt zu einzelnen Familien aus der Umgebung zu suchen, wie es durch die MCAD-Map ermöglicht wird. Auf diese Weise wirst du sehr viel mehr von dem weitestgehend völlig normalen Leben eines vom MCAD-Mangel betroffenen Kindes erfahren, als in einer Diskussionsgruppe, in der es vor allem um problematische Situationen geht. Dabei tritt dann nämlich auch bei noch unerfahrenen Eltern sehr leicht eine gewisse Wahrnehmungsverzerrung ein. Die meisten Teilnehmer erleben nie auch nur die geringsten Probleme mit ihren Kindern – allerdings teilen sie das dann auch nicht der Allgemeinheit mit. Für Tag für Tag hunderte Beiträge in der Art “heute war wieder alles Bestens, mein Kind hatte in der Schule und selbst nachmittags im Sportverein nicht die geringsten Probleme, sondern geht völlig souverän mit seiner Stoffwechselstörung und allem was diesbezüglich wichtig ist um!” ist eine Diskussionsgruppe zum MCAD-Mangel halt der falsche Ort. Stattdessen scheint es durch die von nur vergleichsweise wenigen Familien geschilderten schwierigen Situationen so, als ob alle Kinder mit MCAD-Mangel von einer kritischen Situation in die nächste glitten. Um als MCAD-Neuling wirklich zu verstehen, dass dies keineswegs die alltägliche Realität im Leben mit dem MCAD-Mangel ist, erfordert es schon eine gewisse Erfahrung und Reifung.
Aber auch andere Diskussionen, die sich in einer größeren Gruppe abspielen, können einen Neuling unter Umständen überfordern. Zehn Jahre Forumsleben haben eindrücklich gezeigt, dass so ziemlich alle Stoffwechselzentren unterschiedliche Sichtweisen und somit auch unterschiedliche Empfehlungen bzgl. der den MCAD-Mangel betreffenden Fragestellungen an ihre Patienten weitergeben. Daraus resultiert, dass auf jede vermeintlich einfache Frage, wie es andere Eltern z.B. mit den Nüchternzeiten halten, in 20 Antworten 25 unterschiedliche Empfehlungen zurückgemeldet werden, weil manche Familien selbst in ihren eigenen Kliniken auf keine einheitliche Sichtweise treffen. Sind so viele verschiedene Antworten mit teilweise dann auch noch zwischen einzelnen Personen entstehenden Diskussionen darüber, wer nun mehr Recht hat, wirklich hilfreich? Gilt tatsächlich “je mehr Meinungen, desto besser”? Es wird Rückmeldungen geben, in denen die Nüchternzeiten deutlich kürzer empfohlen werden, als du es selbst kennst, und andere Antworten, in denen die Mahlzeitenpausen für ein Kind im gleichen Alter schon unfassbar lang zu sein scheinen. Kannst du da locker drüberstehen oder wirst du dir Gedanken machen, ob die euch gegenüber gegebenen Empfehlungen vielleicht falsch oder sogar schädlich für dein Kind sein könnten? Wenn zwei oder noch mehr “Experten” ganz unterschiedliche Zeiten nennen, kann doch nur einer von ihnen Recht haben, oder? Und wenn das nicht unser Arzt ist? Hat er überhaupt Ahnung? Können wir ihm denn dann bei den anderen Empfehlungen vertrauen? Sind wir bei ihm oder ihr nicht vielleicht ganz schlecht aufgehoben? Diese Gedanken werden auf jeden Fall kommen − die Frage ist nur, ob und wie du damit umgehen kannst. Hast du selbst denn schon verstanden und auch verinnerlicht, dass es bzgl. mancher Themen, wie z.B. den maximalen Nüchternzeiten − innerhalb eines gewissen und teilweise recht großzügig bemessenen Bereichs ziemlich pillepalle ist, an welche Empfehlungen man sich hält? Gerade die für andere Kinder “geltenden” Nüchternzeiten wurden und werden von den Eltern am häufigsten angefragt und sind dabei das meiner Meinung nach am stärksten überbewertete Thema überhaupt. Im Rückblick hätte ich damals ein paar für alle Teilnehmer verbindliche Forenregeln festlegen sollen und die erste Regel hätte lauten müssen: “Zum Zwecke des psychischen Wohlbefindens aller Teilnehmer vergleichen wir keine Nüchternzeiten − niemals!”. Mal auf uns Erwachsene übertragen − auch wenn man vielleicht manchmal ganz gerne wüsste, was die Kollegen mit der gleichen Stellenbeschreibung so verdienen, hofft man doch dabei insgeheim nur, dass man selbst im Vergleich der Gehälter ganz gut, wenn nicht gar am Besten dasteht. Wie wohl aber würdest du dich selbst bei deinem Arbeitgeber noch fühlen, wenn du dann feststellen müsstest, dass dein Gehalt doch relativ niedrig ist und andere, vielleicht auch noch jüngere oder vermeintlich weniger erfahrene Kollegen, deutlich mehr als du jeden Monat für die gleiche Arbeit bekommen?
Darüber hinaus werden gerade bei der Frage nach den Nüchternzeiten oftmals Äpfel mit Birnen verglichen! Obwohl einzelne Uniklinken schwere und milde MCAD-Ausprägungen immer noch absolut gleich behandeln (und oftmals auch nichts von milden Formen wissen wollen), werden an anderen Stoffwechselambulanzen Kindern mit eindeutig mildem MCAD-Mangel schon sehr früh extrem lange nächtliche Nüchternzeiten zugestanden, die für gleichaltrige Kinder mit schwerer Ausprägung absolut nicht zu empfehlen sind. Zeitenvergleiche machen daher – wenn überhaupt – nur dann Sinn, wenn bei jeder Antwort ganz eindeutig erkennbar ist, wie es um den MCAD-Hintergrund des betreffenden Kindes steht.
In gleicher Weise verhält es sich üblicherweise mit der Frage, ob und wieviel Carnitin gegeben wird, ob in ein oder zwei täglichen Portionen, nur in Krankheitszeiten oder abwechselnd ja nach Ergebnis der letzten Blutuntersuchung. Huch, welche Blutuntersuchung? Unser Arzt hat nichts von irgendwelchen Blutwerten erzählt. Hat er die wichtigen Punkte überhaupt überprüfen lassen? Sollten wir vielleicht schon längst Carnitin geben, und er hat es nur nicht verschrieben, weil er mit den Werten nichts anfangen kann? Kommt Dir das schon ein wenig bekannt vor? Ganz schlimm wird es, wenn dann auch noch Acylcarnitinwerte, bzw. die Menge des bei einzelnen Kindern gemessenen freien Carnitins verglichen werden. Dann kann man sich schnell die Frage stellen, ob denn die Werte des eigenen Kindes jetzt zu niedrig oder zu hoch sind oder warum diese Werte überhaupt nicht gemessen wurden.
Wenn du dich jetzt bereits durch die Inhalte der letzten drei Absätze stark verunsichert fühlst, bist du definitiv noch nicht reif genug für den unvorbereiteten Austausch mit einer größeren Gruppe anderer Betroffener, die alle unterschiedliche Sichtweisen weitergeben. Oft sind diese Sichtweisen – ohne dass die Antwortenden sich dessen tatsächlich bewusst zu sein – noch dazu durch das eigene mehr oder weniger umfangreiche Verständnis der Thematik stark gefärbt und verändert. Längst nicht immer ist das, was jemand als vermeintlich verlässliche Aussagen seiner Ärztin von sich gibt, auch tatsächlich das, was diese wirklich damit sagen wollte. Denke dabei nur an die starke Veränderung der ursprünglichen Botschaft beim “Stille Post”-Spiel bereits durch die erste Person, die diese selbst nur mündlich gehörte Nachricht weitergibt. Außerdem sind die Termine in den Stoffwechselambulanzen oft von Hektik seitens der Ärzte und Aufregung seitens der Eltern geprägt, sodass die meist knappen oder im Gegenteil sehr umfangreich ausfallenden mündlichen Antworten bis zur Rückkehr nach Hause schon halb vergessen sind oder in Teilen falsch erinnert werden (Merke: immer alle eigenen Fragen im Vorfeld schriftlich notieren und die Antworten des Arztes während des Termins wenigstens in Stichworten mitschreiben!). Und viel zu oft vermischen sich in ein und demselben Antwortbeitrag ein paar wenige zutreffende Fakten mit anderen schnell mal ergoogelten und dabei völlig aus dem nicht verstandenen Zusammenhang gerissenen Halbwahrheiten. So wurde ich z.B. mal zu einer nicht nachvollziehbaren Aussage mit bislang völlig unbekannten Fakten aus einer Facebook-Diskussion rückgefragt, weil sich anscheinend eine von der “Mast Cell Activation Disorder” betroffene Person (wohl aufgrund der identischen Abkürzung dieser Krankheit) vermutlich versehentlich in die MCAD-Mangel-Gruppe “eingeschmuggelt” hatte und mit Aussagen und Begriffen um sich warf, die bei einigen Teilnehmern zu größerer Verwirrung führten. Nur so konnte ich mir die an mich weitergeleiteten Aussagen erklären, die überhaupt nicht zum MCAD-Mangel, aber hundertprozentig zur Mastzellenaktivierungsstörung passten. Ob meine diesbezügliche Vermutung damals aber den Weg zurück in die betreffende Facebook-Diskussion gefunden und dort vielleicht zur Aufklärung bzgl. der zuvor gemachten Äusserungen beigetragen hat, ist mir nicht bekannt. Insofern sollte man im Austausch mit vielen anderen Betroffenen auch immer erst ein wenig skeptisch bleiben, wenn plötzlich für alle Teilnehmer neue “Fakten” (z.B. bezüglich Symptomen oder Therapiemöglichkeiten) eingebracht werden. Unter Umständen beziehen sich diese Infos auch einfach nur auf die falsche Krankheit.
Darüber hinaus sind soziale Medien, in denen die Beiträge ganz schnell nach unten oder oben aus dem Sichtbereich rauslaufen, einfach nicht als Informationsspeicher geeignet. Gerade neue Teilnehmer können meist nicht mehr sehen, ob die sie interessierende Fragestellung vielleicht schon öfters diskutiert wurde und fragen dann halt einfach nochmal nach. Daher werden die gleichen Themen in kurzen Zeitabständen wieder und wieder durchgekaut.
Was sind das hauptsächlich für Themen, auf die du unter den aktuellen Beiträgen stoßen wirst? Da die sozialen Medien, wie gerade beschrieben, nicht als Informationsspeicher taugen, sind es Themen, die die Teilnehmer jetzt gerade ganz akut beschäftigen. Als da wären? Nun, das ist nicht viel anders als in den täglichen TV-Nachrichten. Dort geht es um Kriege, Krisenherde, Naturkatastrophen, Ausbreitung von Krankheiten, Flugzeugabstürze, Verbrechen, Politik und Skandale. Damit lockt man die Menschen vor den Fernseher und mit genau solchen Themen rechnet man doch heutzutage fast nur noch. Damit die zur Verfügung stehenden knappen 15 Minuten Sendezeit nicht nur mit schlimmen Dingen gefüllt werden, wird am Ende noch für 15 Sekunden von einem niedlichen Tierbaby berichtet, das in irgendeinem Zoo geboren wurde, oder Bilder von ein paar Krokussen in irgendeinem Garten gezeigt, die darauf hinweisen, dass der Frühling langsam wiederkommt. Dabei passieren auf der Welt jeden Tag unfassbar viele gute und schöne Dinge, aber diese sind in Punkto Nachrichten nun mal nicht zielgruppenrelevant. Es geht hauptsächlich um Themen, über die sich die Zuschauer aufregen oder Sorgen oder sogar Ängste machen können. So ähnlich verhält es sich − wenn auch nicht mit Absicht − wenn sich eine größere Anzahl von Leuten mal gerade schnell gegenseitig mitteilen möchte, was sie aktuell in Bezug auf den MCAD-Mangel beschäftigt. Die Themen sind also vor allem: Krankheiten, schwierige Nahrungsaufnahme, Krankheiten, Verabschiedungen zum vorsorglichen Krankenhausaufenthalt, Krankheiten, Ärger mit der Kita, der Schule oder den ignoranten Stoffwechselärzten, noch mehr Krankheiten und immer wieder werden gerade noch eben verhinderte oder bereits lange zurückliegende Entgleisungen thematisiert. Auf Erfolgsgeschichten und tolle Entwicklungsschritte der Kinder wirst du im schriftlichen Austausch mit gleichzeitig vielen Eltern, wenn überhaupt, dann nur sehr selten stoßen.
Ein letzter wichtiger Punkt, dessen du dir im Voraus bewusst sein solltest, ist der, dass dir in einem großen Teilnehmerkreis vor allem viele “neue” MCAD-Eltern (in ihren ersten 3 bis 4 Jahren mit dem MCAD-Mangel) antworten werden, da diese, aufgrund ihrer Unerfahrenheit, einfach noch das stärkste Interesse und den größten Bedarf an gegenseitigem Austausch haben. Das war schon hier im Forum über die Jahre hinweg sehr deutlich zu beobachten. Bei allen “alten Hasen” stellt sich nach einiger Zeit des Umgangs mit dem MCAD-Mangel eine gewisse Routine ein. Der MCAD-Mangel des Kindes tritt im täglichen Leben mehr und mehr in den Hintergrund und in gleichem Maße lässt auch das Interesse an der Beteiligung in Diskussionsgruppen nach – zumindest bei denjenigen Fragen, auf die sie bereits dutzende Male die gleiche Antwort gegeben haben, also zu den Standardthemen wie Nüchternzeiten, Carnitingaben, Maltodextrin, Ernährung im Krankheitsfall, wann in die Klinik, usw… Selbstverständlich gibt es trotzdem immer mal Antworten von Teilnehmern, die wirklich bei dem, was sie schreiben, auf eine langjährige Erfahrung zurückgreifen können, aber die meisten Antworten werden von neu betroffenen Eltern gegeben, von denen die wenigsten schon die nun mal notwendige Zeit aufwenden konnten, die es halt braucht, um sich intensiver mit dem MCAD-Mangel auseinander zu setzen und die Zusammenhänge zu verstehen. Wenn dann − mit selbstverständlich besten Absichten − eigene Vermutungen oder auch nur halb verstandene Empfehlungen der eigenen Ärzte als vermeintlich gut belegte Ratschläge in großer Zahl und oftmals einander widersprechend weitergegeben werden, können darin die wirklich guten Empfehlungen der wenigen sich noch beteiligenden Eltern mit umfassendem Erfahrungsschatz untergehen. Natürlich − und dass ist das unumstößliche Prinzip jeder Diskussionsgruppe, sei es in Foren oder auf Facebook − hat jeder Teilnehmer in gleicher Weise die Möglichkeit und auch das Recht auf die Weitergabe seiner eigenen Sichtweise. Dessen solltest du dir aber nun mal im Voraus bewusst sein, denn wirklich für sich selbst herausfiltern, wessen Antworten qualitativ hochwertig sind und wer auf der anderen Seite einfach nur zu allen Themen mit schnell ergoogelten Informationen antwortet, ohne diese tatsächlich verstanden und hinsichtlich ihrer Relevanz bewertet zu haben, kann man erst, wenn man sich selbst schon etwas besser mit dem Thema auskennt.
Das sind die Fakten! So war es zur Zeit des Forums, und so wird es mir auch heute noch immer mal wieder aus den Reihen der Facebook-Teilnehmer geschildert. Du musst selbst überlegen, wie sehr du nach dem anfangs sicher vorhandenen Schock über den MCAD-Mangel inzwischen schon wieder psychisch gefestigt bist, um die regelrechte Überschwemmung mit vielen unterschiedlichen Meinungen zu ertragen. Im besten Fall schaffst du es, dir aus den Diskussionen die “Informations-Rosinen” herauszupicken und den Rest auszublenden. Diese Unterscheidung treffen zu können erfordert allerdings schon ein wenig eigene Erfahrung und vor allem ein schon etwas umfangreicheres eigenes Verständnis über die Hintergründe und “Mechanismen” des MCAD-Mangels. Ansonsten kann es aber auch passieren, dass du durch die Masse an unterschiedlichen Sichtweisen überfordert und extrem verunsichert wirst. Dutzende Teilnehmer des früheren Forums haben mich schon wenige Wochen nach ihrer Anmeldung, teilweise auch nur nach Tagen, um die Löschung ihres Accounts gebeten, weil sie es nicht mehr ertragen konnten, aufgrund der vielen unterschiedlichen Empfehlungen der anderen Teilnehmer ständig im Zweifel zu sein, ob sie es denn mit ihrem Kind überhaupt richtig machen. Mit vielen von ihnen hatte ich danach aber noch über Jahre hinweg außerhalb des Forums Kontakt, um ihnen die dann doch immer mal neu aufkommenden Fragen zu beantworten.
Meine persönliche Empfehlung: versuche die andern Eltern, bzw. ihre Charaktere über die zur Verfügung stehenden sozialen Medien ein wenig kennenzulernen, um dann gezielt mit einzelnen Personen Kontakt aufzunehmen, deren Sichtweise du für vernünftig und mit deiner eigenen als einigermaßen kompatibel empfindest. Es bringt überhaupt nichts, sich näher mit denjenigen zu befassen, die vieles ganz anders machen, als es euch selbst seitens eurer Ärzte empfohlen wurde. Gib nicht zu viel darauf, dass du in einer größeren Gruppe möglichst viele Rückantworten bekommst. Gerade bei Themen, die von allen Eltern oder auch Stoffwechselärzten nach Lust und Laune gehandhabt werden (beim MCAD-Mangel hat tatsächlich so ziemlich jede Klinik ihre ganz eigene Sicht- und Vorgehensweise!), bringt das eher Verunsicherung als Klarheit. Lass Dich nicht davon verrückt machen, dass es in fast allen Beiträgen hauptsächlich um Probleme geht. In Wirklichkeit machen diese scheinbar so allgegenwärtigen und ständigen Probleme im tatsächlichen Leben jeder einzelnen Familie nur einen winzig kleinen Teil aus, aber überall dort, wo sich Betroffene in einer sie verbindenden Gruppe zusammenschließen − sei es Forum, WhatsApp oder Facebook − geht es fast ausschließlich um diesen winzig kleinen Teil und durch die Menge der Beiträge erscheint er dann doch wieder wie ein riesiger Berg. Das ist nun mal das Wesen von Diskussionsgruppen im Internet. Sei Dir also bewusst, dass dich all dies erwarten wird und dann überlege dir, ob du es jetzt schon ertragen kannst, oder ob du erst noch ein wenig mehr Sicherheit im Umgang mit dem MCAD-Mangel erlangen solltest, bevor du dich auf den Austausch mit einer größeren Gruppe anderer Eltern einlässt. Die zu Zeiten des Forums entstandene “Es gibt Eltern-Liste” im Artikel “Lohnende Gedanken” kann dich auch noch ein wenig darauf vorbereiten.![]()