Kommt Dir das bekannt vor?
Wieso hat gerade unser Kind einen MCAD-Mangel? Wie konnte es bloß dazu kommen? Wenn es doch durchschnittlich nur bei einem von zehn- bis fünfzehntausend Kindern gefunden wird, warum dann gerade bei meinem Kind?
Die Frage nach dem “Warum?” und der unerklärlichen Herkunft des MCAD-Mangels werden sich vermutlich alle Eltern in der ersten Zeit nach der Diagnose vermehrt stellen. Die Nachricht kommt im Normalfall völlig unerwartet und muss erst einmal verarbeitet werden.
Vielleicht können Dir dabei ein paar Denkanstöße helfen, die in dem Artikel “Lohnende Gedanken” behandelt werden. An dieser Stelle hier geht es allerdings zunächst mal um die medizinischen, bzw. biologischen Hintergründe.
Wo kommt dieser Gendefekt so plötzlich her?
Das Auftreten eines genetischen Defekts, wie dem, der die Bildung des MCAD-Enzyms stört, kann zwei unterschiedliche Gründe haben. Entweder handelt es sich um eine spontane Mutation des Erbguts, die rein zufällig auf genau diesem Chromosom und genau diesem Gen aufgetreten ist, oder aber die Mutation wurde von den Eltern an das Kind vererbt.
Aber Moment mal! Bei uns Eltern ist in der Kindheit nie etwas passiert! Wir hatten auch die ganzen üblichen Infekte und ein Dutzend Mal Brechdurchfall, und trotzdem haben wir nie so etwas wie eine Stoffwechselkrise erlebt. Auch die eine oder andere ungesunde Radikaldiät haben wir problemlos überstanden.
Das sind verständliche Einwände, aber trotzdem können Kinder einen MCAD-Mangel von ihren Eltern vererbt bekommen, obwohl diese selbst keinen haben, bzw. es bei ihnen niemals bemerkt wurde. Wie hätte er auch festgestellt werden sollen, wird der Schnelltest auf die auch den MCAD-Mangel einschließenden zusätzlichen Stoffwechselstörungen doch erst seit wenigen Jahren bundesweit durchgeführt. Rein rechnerisch betrachtet hat im Durchschnitt auch höchstens eins von 100-120 der MCAD-Elternteile selbst einen MCAD-Mangel von seinen eigenen Eltern vererbt bekommen. Der tatsächliche Anteil ist vermutlich deutlich niedriger, denn da der MCAD-Mangel während unserer eigenen Kindheit wie erwähnt noch nicht frühzeitig getestet werden konnte und somit die bei einer Entgleisung auftretenden Symptome oft nicht richtig eingeschätzt wurden, sind sicher einige der damaligen davon betroffenen Kinder, die heute im Erwachsenenalter und selbst Eltern wären, während einer frühen Krise bereits verstorben.
Die Übertragung des MCAD-Gendefekts durch Vererbung scheint nach den Erfahrungen der letzten Jahre der übliche Weg zu sein. Die spontane Mutation kann zwar rein theoretisch auch vorkommen, ist aber extrem selten und führt dabei wiederum mit großer Wahrscheinlichkeit dazu, dass die betreffende Person nur Träger (“Carrier”) des Gendefekts ist, aber selbst keinen MCAD-Mangel hat (siehe unten). Natürlich können auch spontane Mutationen später an die eigenen Kinder vererbt und somit über die folgenden Generationen hinweg verbreitet werden.
Um nun den Weg über die Vererbung näher zu betrachten, müssen wir uns an den Biologieunterricht in der Schule erinnern.
Die Vererbungslehre nach Mendel
Jeder Mensch hat in seinen Zellkernen den vollständigen Chromosomensatz in doppelter Ausführung. Die eine Hälfte der Chromosomen mit allen darauf kodierten Merkmalen bekam er von seiner Mutter, die andere Hälfte von seinem Vater vererbt. Bei der Verschmelzung von Eizelle und Spermium entstand eine erste Zelle mit dem doppelten Chromosomensatz, welcher durch die fortschreitende Zellteilung in jede neue Zelle weitergegeben wurde.
Die Chromosomen werden zur einfacheren Unterscheidung der Größe nach durchnummeriert und in 2 Klassen unterteilt. Jeder Mensch hat 44 Autosomen (2 mal 22) und 2 Gonosomen (2 mal 1). Autosomen enthalten die allgemeinen Merkmale, wie z.B. die Augen- oder Haarfarbe, während die beiden Gonosomen das Geschlecht des Menschen und alle daran gekoppelten speziellen Merkmale festlegen. Hier spricht man von X-Chromosom und Y-Chromosom. Eizellen enthalten stets das X-Chromosom, und die Spermien der Männer transportieren entweder ein X- oder ein Y-Chromosom. Treffen in der befruchteten Eizelle zwei X-Chromosomen aufeinander, entwickelt sich daraus ein Mädchen, bei einem X- und einem Y-Chromosom wird daraus ein Junge.
Die meisten Stoffwechselstörungen werden autosomal rezessiv vererbt − so auch der MCAD-Mangel. Autosomal bedeutet einfach nur, dass sich das Merkmal zur Bildung des MCAD-Enzyms auf einem der Autosomen und nicht auf einem Gonosom befindet. Von dem Gendefekt können also Mädchen und Jungen gleichermaßen betroffen werden. Im Gegensatz dazu gibt es auch gonosomale Erbgänge, z.B. die Bluterkrankheit, bei der sich das betreffende Merkmal auf dem X-Chromosom befindet. Von der Bluterkrankheit sind mehr Männer als Frauen betroffen, da Männer nur ein X-Chromosom besitzen, und sich dessen fehlerhafte Anlage somit unmittelbar auswirkt. Frauen können mit ihren zwei X-Chromosomen den geerbten Defekt auf der einen Genkopie durch die intakte zweite Genkopie ausgleichen. Dies ist das Entscheidende bei der rezessiven Vererbung.
Rezessiv versus Dominant
Die 22 von der Mutter erhaltenen Autosomen enthalten genau die gleichen Gene wie die 22 vom Vater beigesteuerten Autosomen, aber die Ausprägung dieser Gene kann sich voneinander unterscheiden. Man bezeichnet die einander entsprechenden, aber nicht unbedingt gleich ausgeprägten Gene als “Allele”.
Bekommt ein Kind z.B. von der Mutter das Allel für blaue Augen und vom Vater das Allel für braune Augen vererbt, wird es selbst braune Augen bekommen, weil das Braun-Allel dominant gegenüber dem untergeordneten (rezessiven) Blau-Allel ist. Das Allel für blaue Augen ist aber nach wie vor in seinen Chromosomen enthalten und kann bei der Fortpflanzung an das eigene Kind weitergegeben werden. Damit ist klar, das jeder Mensch mit blauen Augen zwingend zwei Allele für blaue Augen in seinem Erbmaterial mit sich führen muss und auch nur die Anlage für die Augenfarbe Blau an sein Kind weitergeben kann. Haben beide Eltern blaue Augen, kann das Kind auch nur blaue Augen bekommen und auch nur diese Augenfarbe an spätere eigene Kinder weitergeben. Haben dagegen beide Eltern braune Augen, kann ihr Erbgut trotzdem ein rezessives blau-Allel enthalten, so dass das gemeinsame Kind am Ende möglicherweise doch blaue Augen hat. Dieses 1907 von Davenport für die Vererbung der Augenfarbe aufgestellte 1-Gen-Modell ist nicht ganz zutreffend, da man heute weiß, dass an der Augenfarbe mindestens 3 Gene beteiligt sind, deren Zusammenspiel man noch nicht richtig kennt, aber dieses vereinfachte Modell verdeutlicht den Unterschied zwischen dominanten und rezessiven Allelen trotzdem ganz gut.
Schon 1909 hat William Bateson (Cambridge University) festgestellt, dass dominant vererbte Erkrankungen anscheinend grundsätzlich daher rühren, dass dem menschlichen Körper durch die Auswirkung einer genetischen Mutation irgendeine neue “Zutat” hinzugefügt wurde, z.B. irgendeine körpereigene chemische Substanz, deren Vorhandensein diesen Krankheitseffekt auslöst. Ein Beispiel dafür ist die Huntington-Krankheit, bei der durch das von einem Defekt betroffene Gen Proteine gebildet werden, die wie ein Nervengift schädigend auf das Gehirn des Menschen wirken und es nach und nach und unaufhaltsam zerstören. Es nützt dabei nichts, dass die Proteine, die aus dem vom anderen Elternteil geerbten entsprechenden Gen gebildet werden, diesen schädigenden Effekt nicht haben. Sie können die Wirkung des toxischen Proteins nicht neutralisieren, denn dieses hat eine neue und im Körper normalerweise nicht vorhandene Funktion, gegen die es kein Gegenmittel gibt. Auch wenn dieser Gendefekt also nur heterozygot vorliegt, wirken sich die von ihm verursachten Schäden bei der Protein-Biosynthese immer und in vollem Umfang aus. Dieser Gendefekt, bzw. dessen verursachter Schaden ist somit dominant. Im Gegensatz dazu lässt sich bei rezessiv vererbten Krankheiten beobachten, dass dem Körper durch einen Gendefekt irgendetwas normalerweise vorhandenes genommen wurde. Das Nichtvorhandensein einer bestimmten Substanz, bzw. das Fehlen deren Funktion, löst die rezessiv vererbte Krankheit aus. In beiden Fällen gilt aber der wichtige Grundsatz: “Funktion dominiert über Funktionsmangel!” Wie das zu verstehen ist, wird im folgenden Abschnitt erklärt.
Wie wird der MCAD-Mangel vererbt?
Auch der für den MCAD-Mangel verantwortliche Gendefekt wird ausschließlich rezessiv vererbt. Ist ein Allel mutiert, gibt es im Normalfall immer noch seine “Sicherheitskopie” in Form des entsprechenden Allels auf dem vom zweiten Elternteil beigesteuerten Chromosom. Die ordnungsgemäße Funktion dieses “Wildtyps” ersetzt, bzw. dominiert über den Funktionsmangel des mutierten Allels. Somit kann die betreffende Person trotz einseitigem Gendefekt ein voll funktionstüchtiges MCAD-Enzym in ausreichender Menge bilden. Sie ist nur Anlagenträger, also “Carrier”.
Man darf es sich allerdings nicht so vorstellen, dass bei Carriern das vom MCAD-Gendefekt betroffene Allel überhaupt keine Rolle spielt. Im Körper werden Enzyme bei Bedarf ständig neu aus Aminosäuren zusammengebaut und bei einem irgendwann bestehenden Überangebot bzw. bei Bedarfsende auch wieder in ihre Bestandteile zerlegt, um daraus dann neue Enzyme, bzw. ganz allgemein Proteine zu bauen. Der Bauplan für die benötigten Enzyme wird – mal ganz vereinfacht dargestellt – als Negativ-Abdruck aus der in den Chromosomen enthaltenen DNA ausgelesen. Wenn MCAD-Enzyme benötigt werden, wird dazu der entsprechende Abschnitt (also das MCAD-Gen) auf einem der beiden DNA-Stränge gesucht und davon ein Abdruck gemacht. Dieser Abdruck wird dann von einer “Proteinfabrik” in Dreierblöcken abgetastet, und je nach Muster fügt sie die unterschiedlichen Aminosäuren zu einer langen Kette zusammen, bis das Ende des DNA-Abdrucks oder eine Stop-Markierung erreicht ist. Von welcher der beiden existierenden Genkopien (eine mit MCAD-Gendefekt und eine ohne) der Abdruck gemacht wird, ist vollkommen zufällig. Es werden deshalb von den Proteinfabriken genau so viele MCAD-Enzyme mit Funktionsstörung gebildet, wie intakte Enzyme. Reicht die Menge der funktionsfähigen Enzyme nicht aus, um den aktuellen Bedarf zur Aufspaltung der Fettsäuren zu decken, werden weiter MCAD-Enzyme gebaut. Es existiert dann zwar genaugenommen ein absolutes Überangebot an MCAD-Enzymen zur Erledigung dieser Aufgabe, aber da nur die Hälfte von ihnen funktioniert, wird der Bedarf dadurch genau gedeckt. Es ist also nicht so zu verstehen, dass bei rezessiv vererbten Krankheiten nur die “gute” Genkopie verwendet und die beschädigte Genkopie quasi ausgeblendet wird. Die von der intakten Genkopie gebildeten Proteine/Enzyme reichen einfach aus, um ihre Aufgabe zu erledigen und schaffen das mit weg, was die gleichzeitig genau so vielen vermurksten Proteine/Enzyme nicht leisten können.
Damit ist verständlich, weshalb sich der MCAD-Gendefekt bereits über Generationen innerhalb einer Familie vererbt haben kann, ohne dass jemals irgendwelche Auswirkungen davon zu bemerken waren. Alle Personen waren nur einseitige Carrier, also Überträger eines mutierten Allels, aber nicht selbst am MCAD-Mangel erkrankt, da sie vom anderen Elternteil immer ein normales Allel beigesteuert bekamen.
In irgendeiner Generation kann es aber passieren, dass zufälligerweise sowohl der Vater als auch die Mutter Träger des Defekts sind und beide ausgerechnet jeweils den von dem Defekt betroffenen Chromosomenanteil an ihr Kind weitergeben. Wenn also weder das väterliche noch das mütterliche Allel in der Lage sind, ein voll funktionsfähiges MCAD-Enzym zu bilden, hat das Kind einen echten MCAD-Mangel. Die Wahrscheinlichkeit für das Eintreten dieses Falles liegt unter den genannten Voraussetzungen (beide Eltern sind Carrier) bei 25%.
Bekommt es dagegen nur von einem der beiden Carrier-Elternteile ein mutiertes Allel vererbt (50%-Chance), wird es selbst zum Carrier, ist aber selbst nicht von einem MCAD-Mangel betroffen. Bei ihm liegt der Gendefekt “heterozygot”, also nur auf einem Allel vor.
Bekommt es von beiden Elternteilen das Chromosom mit dem nicht-mutierten Gen vererbt (25%-Chance), trägt das Kind den MCAD-Gendefekt nicht in seinem Erbmaterial und kann ihn somit auch nicht an die eigenen Kinder weitergeben.
Unterscheidung der Begriffe MCAD-Defekt und MCAD-Mangel, wie sie auf diesen Seiten hier verwendet werden
Anhand der vorausgegangenen Ausführungen wurde vielleicht schon verständlich, warum in diesem und den weiteren Artikeln immer deutlich zwischen den Begriffen “MCAD-Defekt” und “MCAD-Mangel” unterschieden wird, auch wenn dies in den deutschen Uni-Kliniken längst nicht so sorgfältig gehandhabt wird. Alle Carrier, zu denen auch wir als Eltern eines Kindes mit nachgewiesenem MCAD-Mangel zwingend gehören, tragen ebenfalls einen MCAD-Defekt in den Genen, der jedoch nicht zum Entstehen eines MCAD-Enzym-Mangels führt. Dieser resultiert erst aus dem Vorliegen eines doppelten MCAD-Defekts, wenn sowohl der von der mütterlichen, als auch der väterlichen Seite geerbte Chromosomenanteil ein mutiertes, also “defektes” Gen aufweist.
Der MCAD-(Gen-)Defekt ist gewissermaßen die Ursache und der bei doppelter Ursache entstehende MCAD-Mangel die Wirkung.
In einigen den MCAD-Mangel behandelnden, hauptsächlich deutschen Informationen und vor allem auch im Sprachgebrauch vieler deutscher Ärzte werden diese beiden Begriffe jedoch nicht sauber auseinander gehalten, sondern gleichbedeutend verwendet und mit der Behauptung, das sei doch dasselbe, ganz nach Belieben durcheinander geworfen. In englischsprachigen Artikeln − selbst wenn diese von deutschen Medizinern verfasst wurden − wird bzgl. dieser Stoffwechselstörung dagegen nie von MCAD-defect, -failure, -damage oder -fault (dies wären mögliche Übersetzungen für einen “Defekt”), sondern ausschließlich von MCAD-Deficiency (Mangel, Fehlmenge, Defizit, Unzulänglichkeit) gesprochen. Möglicherweise rührt die Unsitte, in einigen deutschen Kliniken von MCAD-Defekt zu sprechen daher, dass die englische Bezeichnung MCAD-Deficiency auch oft als MCADD abgekürzt wird. Um dieses letzte “D” wieder mit einen deutschen Begriff zu füllen, bietet sich Defekt nun mal eher an als Mangel. Die offizielle Bezeichnung dieser Stoffwechselstörung ist jedoch MCAD-Deficiency, bzw. MCAD-Mangel!
Man kann sich nun die Frage stellen, wo überhaupt das Problem liegt. Der eine Begriff ist doch so gut wie der andere… Die Erfahrungen der Vergangenheit haben jedoch schon sehr deutlich gezeigt, dass das Durcheinanderwerfen dieser Begriffe zu anhaltenden Missverständnissen führen kann − nicht nur bei den Eltern eines Kindes mit MCAD-Verdacht, sondern auch bei den behandelnden Ärzten, die oftmals über nur sehr geringe Hintergrundkenntnisse zum MCAD-Mangel verfügen.
Da man nämlich nie weiß, ob und wie ernsthaft die einzelnen Institutionen zwischen diesen Begriffen unterscheiden, führt dies möglicherweise zu einiger Verwirrung. Meldet das Screeningzentrum in dem ersten Befund z.B. den Verdacht auf einen “MCAD-Defekt”, kann dies zwar einen angenommenen und behandlungsbedürftigen MCAD-Mangel bedeuten, möglicherweise meint man es aber auch tatsächlich genau so, wie man es geschrieben hat und vermutet aufgrund relativ schwach erhöhter Werte auch nur das Vorliegen eines heterozygoten Gendefekts, der zwar noch genau abgeklärt werden sollte, sich aber höchstwahrscheinlich nicht in Gestalt eines MCAD-Mangels auswirken wird. Gegebenenfalls sollte man als Eltern mit Hilfe des Kinderarztes einfach noch mal genau nachfragen, was mit dem im Befund genannten Begriff “MCAD-Defekt” genau gemeint wurde.
Zu besonders schweren Missverständnissen führt das Durcheinanderwürfeln dieser Begriffe dann, wenn den behandelnden Ärzten das Prinzip der rezessiven Vererbung selbst nicht so ganz klar zu sein scheint. Es gab nämlich schon mehrere Fälle, in denen bei einem Kind mit im Screening aufgekommenem MCAD-Verdacht, trotz vollständiger Sequenzierung nur ein heterozygoter MCAD-Gendefekt gefunden wurde – ebenso bei einem Elternteil, beim anderen aber nichts. Statt aus diesem Ergebnis – in Kombination mit dem bei ihm nur knapp über den Grenzwerten liegenden Screeningwerten – den korrekten Rückschluss zu ziehen, dass das Kind dann nur heterozygoter Anlagenträger (ohne MCAD-Mangel!) sein kann, schlussfolgerte der behandelnde Arzt gegenüber den Eltern: “Dann haben Sie also das gleiche wie ihr Kind, nur dass es bei ihrem Kind eine schwere Form ist, und bei Ihnen was mildes.” Wenn jemand so etwas behauptet, dann hat er die beim MCAD-Mangel zur Auswirkung kommende rezessive Vererbung nicht verstanden, denn der bei dem Kind möglicherweise vorliegende MCAD-Mangel ist immer die Summe aus den vererbten Genen beider Eltern, niemals von nur einem einzelnen Elternteil. Umgekehrt können die Eltern unmöglich den gleichen MCAD-Mangel wie das Kind haben, denn dazu fehlt ihnen einfach der vom jeweils anderen Elternteil beigesteuerte Chromosomenanteil, der den MCAD-Mangel des Kindes erst zu einem solchen machte. Wenn der behandelnde Arzt aber selbst nicht verstanden hat, was der Unterschied zwischen einem heterozygoten MCAD-Gendefekt und einem durch einen homozygotem oder compound heterozygotem Gendefekt entstehenden MCAD-Mangel ist, dann kommt es leider zu solchen falschen Auskünften gegenüber den Eltern, die dadurch dann vollends verwirrt werden. (Wichtiger Hinweis an dich: Wenn du die Aussagen eines Arztes z.B. zu den Befunden nicht nachvollziehen kannst, liegt es nicht immer ausschließlich an dir und deiner medizinischen Unkenntnis – manchmal ist das, was du von deinem Arzt gesagt bekommst auch schlicht und einfach falsch und du bist daher zu Recht verwirrt.) Die einzige Möglichkeit, in der ein Elternteil wirklich genau “das Gleiche” wie das Kind haben könnte, wäre die, dass der andere Elternteil selbst keinen Gendefekt beigesteuert hat, und somit Mutter und Kind, oder Vater und Kind beide definitiv nur Carrier sind. Dann haben sie aber beide keinen MCAD-Mangel! Selbst wenn Mutter oder Vater selbst einen MCAD-Mangel von ihren beiden Eltern vererbt bekommen haben sollten, kann das Kind nicht das Gleiche wie dieser eine Elternteil haben, denn auch der selbst vom MCAD-Mangel betroffene Elternteil gibt immer nur eine seiner beiden defekten Genkopien weiter, niemals aber beide zusammen.
Ebenso ist die schon mehrfach gegenüber den früheren Forumsmitgliedern geäusserte Behauptung, das Kind würde seinen “MCAD-Defekt” später einmal an seine eigenen Kinder weiter vererben, nicht zutreffend − zumindest dann nicht, wenn der betreffende Arzt mit diesem Begriff mal wieder den MCAD-Mangel meinte, was leider auch meistens der Fall ist. Auch ein Kind mit MCAD-Mangel, das also über zwei defekte Genkopien verfügt, kann immer nur eines dieser beiden defekten Gene (bzw. Allele) an seine eigenen Kinder weitergeben. Deren zweite Genkopie wird vom anderen Elternteil beigesteuert, also entweder im Spermium des Vaters oder der Eizelle der Mutter. Da nur etwa jede 60. Person in Deutschland Träger eines einzelnen defekten MCAD-Gens ist, sind die Chancen mit über 98% doch relativ groß, dass auch die Kinder eines selbst vom MCAD-Mangel betroffenen Menschen nur Träger sein werden. MCAD-Betroffene vererben später also niemals den MCAD-Mangel, sondern nur dessen Anlage, also einen einzelnen MCAD-Gendefekt, der für sich allein genommen erst einmal keine Bedeutung hat.
Solche schon mehrfach in den Reihen der Forumsmitglieder aufgetretenen Missverständnisse hätten sich nahezu problemlos vermeiden lassen, wenn man die Begriffe MCAD-Mangel für die Stoffwechselstörung und MCAD-Gendefekt ordentlich auseinander halten, und den missverständlichen und weder das eine noch das andere − oder auch beides − beschreibenden Begriff MCAD-Defekt aus dem Sprachgebrauch schnell wieder streichen würde.
Arten von Mutationen
Polymorphismen
Nicht alle genetischen Veränderungen des Erbguts werden als Mutation bezeichnet. Es kommt dabei auf die Verbreitungshäufigkeit an. Als Polymorphismen bezeichnet man in der Medizin diejenigen genetischen Abweichungen vom Wildtyp, für die in der Bevölkerung eine relativ große Verbreitung nachgewiesen wurde. Gemäß Definition muss eine solche Abweichung bei mehr als 1% der Bevölkerung vorkommen, damit von einem Polymorphismus gesprochen werden kann. Abweichungen, die bei weniger als 1% der Bevölkerung auftreten, werden dagegen als Mutationen bezeichnet. Polymorphismen haben meist keinen Krankheitswert, bzw werden ihre Auswirkungen aufgrund der hohen Verbreitungshäufigkeit als normal angesehen.
Stille Mutationen
Zu den vorgenannten Polymorphismen werden auch die sogenannten “stillen” Mutationen gezählt. Es gibt nämlich 64 (4 hoch 3) verschiedene Kombinationen von Tripletts aus den vier verschiedenen Basen, die jedoch für nur insgesamt 20 verschiedene Aminosäuren kodieren. Die meisten dieser Aminosäuren werden somit von mehr als einer möglichen Dreierkombination der Basen festgelegt. Bei einer stillen Mutation wird zwar durch den Austausch einer Base in der DNA ein anders lautendes Triplett gebildet, welches jedoch für die gleiche Aminosäure codiert, wie die ursprüngliche Basensequenz. Aus diesem Grund haben stille Mutationen meist keinen eigenen Krankheitswert, sie könnten im Zusammenspiel mit anderen Mutationen auf dem gleichen Allel aber möglicherweise doch eine Bedeutung haben. So hat eine Untersuchung aus dem Jahr 2007 den Schluss gezogen, dass beispielsweise die durch die Mutation c.362c>t hervorgerufenen negativen Splicingeffekte bei gleichzeitigem Vorliegen der stillen Mutation c.351a>c erstaunlicherweise weitgehend vermieden wurden. Warum das so ist, ist unklar. Möglicherweise ist es auch einfach nur Zufall gewesen, da man sich vorstellen kann, dass es aufgrund der ohnehin schon extremen Seltenheit bestimmter Mutationen dann auch nur eine sehr kleine Vergleichsgruppe gibt, in der diese beiden Mutationen gemeinsam auftreten. Meistens sind die von solchen genetischen Konstellationen betroffenen Personen blutsverwandt (Eltern-Kinder oder Geschwister) und aufgrund des nahen Verwandschaftsverhältnisses könnten auch ganz andere genetische Einflussgrößen zu den gemachten Beobachtungen geführt haben. Festzuhalten bleibt daher, dass stille Mutationen üblicherweise keinen Krankheitswert haben.
Missense Mutations
Die im ACADM-Gen, sowie in jedem anderen Gen auftretenden Mutationen können unterschiedlicher Ausprägung sein. Am häufigsten sind die sogenannten missense mutations. Diese bewirken, dass durch den Austausch eines einzelnen Nukleotids im DNA-Strang ein falscher 3er-Code für die Einfügung einer Aminosäure in das aus diesem Bauplan zu erzeugende Protein entsteht. Jede der Aminosäuren hat jedoch eine spezielle dreidimensionale Molekül-Struktur, die von ihrem Anfangs- zu ihrem Endpunkt in einem bestimmten Winkel abknickt. Wird nun eine andere Aminosäure eingefügt, entsteht an dieser Stelle ein falscher Richtungswechsel und die lange Aminosäurenkette faltet sich nicht mehr zu der korrekten dreidimensionalen Struktur zusammen, die erforderlich ist, um dem finalen Protein seine als Enzym zu erfüllende Funktion zu ermöglichen. Je nach der Position der falschen Aminosäure ist das entstehende MCAD-Enzym mehr oder weniger stark eingeschränkt funktionsfähig. Die in westlichen Ländern am häufigsten gefundene Mutation K329E ist eine solche missense mutation und führt zur Produktion eines instabilen Proteins mit stark eingeschränkter Aktivität (Anmerkung: in einem früheren Forschungsbericht wurde die Aussage getroffen, dass die K329E-Mutation zu keinem nennenswerten Aktivitätseinbruch des gebildeten Enzyms führe, sondern dieses lediglich bei erhöhten Temperaturen, also bei Fieber, schnell instabil würde. Mittels der heute sehr viel genaueren Verfahren zur Aktivitätsmessung des MCAD-Enzyms hat sich aber deutlich gezeigt, dass diese Mutation zur Bildung von Enzymen mit so gut wie keiner Restaktivität in Bezug auf die Verarbeitung mittelkettiger Fettsäuren führt.) Da die gesamte Mutation darin besteht, dass an einem bestimmten Punkt im Code eine einzelne Base durch eine andere Base ersetzt wird, bezeichnet man diese Art von Mutationen auch als “Punktmutationen”.
Nonsense Mutations bzw. Stop-Mutationen
Jede Gensequenz, auch die für die Bildung des MCAD-Enzyms verantwortliche, beginnt mit einem genau definierten Start-Codon (dem Basentriplett ATG, welches für die Aminosäure “Methionin” codiert, die anzeigt, dass ab hier die Beschreibung für ein neues Protein beginnnt) und endet mit einem von drei möglichen Stop-Codons (TAA, TAG, TGA). Durch eine genetische Mutation kann nun z.B. aus einem irgendwo in dieser Sequenz angeordneten “harmlosen” Glutamin-Codon (CAA) durch den Austausch von C nach T ein solches Stop-Codon (TAA) werden. Das gleiche kann auch einem Lysin-Codon (AAA) passieren, wenn dort ein Austausch von A nach T stattfindet. Eine nonsense mutation führt zur Erzeugung eines solchen vorzeitigen Stop-Codons (oder Nonsense-Codons) im genetischen Code und damit wird die Bildung des betreffenden Proteins weitgehend verhindert. Gewissermaßen wird die Bauphase beendet, bevor das Enzym fertig gebildet wurde.
Deletions und Insertions
Deletions löschen den DNA-Code an einer bestimmten Stelle. Insertions fügen stattdessen ein oder mehrere zusätzliche Basen in den Code ein. In beiden Fällen verschiebt sich das Leseraster ab der betreffenden Position bis zum Ende des Gens um eine, oder mehrere Stellen. Dies ist insofern problematisch, da immer drei Basen zusammen ein Basentriplett bilden, mit dem eine der 20 möglichen Aminosäuren codiert ist, aus denen sich das Protein zusammensetzt. Fehlt nun z.B. durch eine Deletion eine dieser Basen, rutscht eine Base aus dem nächsten Dreierblock nach, so dass ab der betroffenen Stelle alle folgenden Basentripletts für eine falsche Aminosäure codieren. In so einem Fall entsteht meist ein völlig falsches Protein und daraus resultierend auch ein weitgehend nutzloses MCAD-Enzym. Im günstigsten Fall werden bei einer Deletion oder Insertion gleich eine durch drei teilbare Anzahl an Basen gelöscht oder eingefügt, sodass alle weiteren Aminosäuren trotzdem korrekt codiert werden. Das entstehende Protein ist dann zwar auch fehlerhaft, das Enzym kann aber möglicherweise noch einen deutlichen Restnutzen aufweisen. Die Verschiebung des Leserasters um ein oder zwei Stellen führt meist zu einem kurz nach der betreffenden Position entstehenden Stop-Codon, an dem die Verarbeitung abbricht, so dass gar nicht erst ein zuviel Murks anrichtender Proteinabschnitt entstehen kann.
Splicing Mutations
Die meisten bekannten Mutationen liegen auf den nicht für die Codierung des Proteins zuständigen Zwischenbereichen des Gens, den sogenannten Introns. Im DNA-Strang werden die das Protein beschreibenden (codierenden) Abschnitte, die sogenannten Exons, an mehreren Stellen durch diese Introns unterteilt. Insgesamt bestehen ca 45% des menschlichen Genoms aus diesen nicht für die Erzeugung der Proteine/Enzyme benötigten Introns. Während der Bildung der mRNA aus der anhand der DNA erzeugten RNA (Ribonukleinsäure) fallen diese Introns weg, und die den Code enthaltenden Exons verbinden sich miteinander. Dieser Vorgang wird als Splicing oder Spleissen bezeichnet. Daher haben auf den Introns liegende Genmutationen, von denen es im menschlichen Genom eine große Menge gibt, normalerweise keine wesentliche Bedeutung, da sie in den gebildeten Enzymen ohnehin nicht mehr enthalten sind. Einzelne dieser Mutationen können jedoch zu einem falschen Ablauf des Splicings führen, so dass in der entstehenden mRNA z.B. eines der an das betreffende Intron angrenzenden Exons fehlt, oder sich die Exons in irgendwie gearteter fehlerhafter Weise aneinanderreihen. Manche Intron-Mutationen zerstören auch die Anfangs- oder Ende-Kennungen der Intron-Abschnitte, so dass Teile dieser eigentlich zu entfernenden Codesequenzen mit in das letzten Endes gebildete Protein eingearbeitet werden. Dies kann z.B. vollständig funktionslose, aber in manchen Fällen auch Proteine mit sogar schädlichen Auswirkungen zur Folge haben.
Das folgende eingebettete Video entstand als Arbeit für das Bio-Abi (genauere Angaben sind auf der Youtube-Seite leider nicht enthalten) und zeigt sehr schön und einfach erklärt, wie aus den in den Zellkernen enthaltenen Chromosomen mit den darauf enthaltenen Genen in mehreren Arbeitsschritten Proteine produziert werden. Aus dem MCAD-Gen entstehen auf diese Weise die Proteine, die dann als MCAD-Enzyme bei der Aufspaltung der Fettsäuren tätig werden. In diesem Video wird auch kurz der Arbeitsschritt des Splicings erklärt.
Bekannte Mutationen
Die am weitesten verbreitete und in der Vergangenheit häufig nach einer Stoffwechselentgleisung gefundene MCAD-Mutation, in der Literatur oft als “prävalent” (vorherrschend) bezeichnet, trägt die Bezeichnung K329E. Eine andere Bezeichnung, die den erfolgten Basenaustausch beschreibt, lautet c.985A>G. In englischen Artikeln wird diese Mutation oft auch in der alten Nomenklatur K304E beschrieben, bei der mit der Zählung ein paar Stellen weiter hinten begonnen wird. Es handelt sich aber um die gleiche Mutation. Sie wurde bislang bei etwa 80% der von einer Stoffwechselentgleisung betroffenen Patienten im Rahmen der daraufhin durchgeführten molekulargenetischen Untersuchung homozygot gefunden. Homozygot bedeutet, dass die Mutation auf beiden Chromosomen zu finden ist, es in diesem Fall also keine intakte Wildtyp-Sicherheitskopie gibt. In weiteren 18% der erst nach einer Stoffwechselentgleisung gefundenen Fälle liegt K329E compound heterozygot mit einer anderen Mutationen vor. Compound heterozygot bedeutet, dass beide Allele unterschiedliche Mutationen aufweisen, die aber jede für sich die ordnungsgemäße Bildung des MCAD-Enzyms einschränken. Mehr dazu, wie diese Prozentangaben genau zu verstehen sind, ist im Info-Artikel “Die Diagnose des MCAD-Mangels” erläutert. Die Carrier-Häufigkeit für eine den MCAD-Mangel auslösende Mutation innerhalb der deutschen Bevölkerung, liegt grob geschätzt irgendwo im Bereich von 1/60, was bedeutet, dass etwa jeder sechzigste Deutsche − größtenteils unwissend − eine solche Mutation in wenigstens einem seiner Chromosomen mit sich führt und möglicherweise auch an seine Kinder weitergibt. Bislang wurden über 340 verschiedene und teilweise sehr seltene Mutationen des ACADM-Gens identifiziert, von denen aber längst nicht alle zu einer schweren Funktionsstörung des aus dem betreffenden Gencode gebildeten Enzyms führen.
Es gibt auch untersuchte Fälle, in denen auf ein und demselben Chromosom gleich zwei Mutationen in einem Allel auftreten (“cis-Stellung”), das Allel auf dem vom anderen Elternteil beigesteuerten Chromosom aber den Wildtyp aufweist. Somit liegt bei diesen Patienten kein MCAD-Mangel vor und man kann nicht feststellen, wie sich diese beiden Mutationen auswirken würden, befänden sie sich nicht gemeinsam auf einem, sondern verteilt auf beiden Chromosomen (“trans-Stellung”). Im Rahmen der Gensequenzierung werden dann u.U. zwar beide Mutationen gefunden, in manchen Fällen kann dann aber erst die genetische Untersuchung der Eltern wirklich Klarheit bringen. Werden beide Mutationen auch bei einem der Elternteile gefunden, während der andere Elternteil diesbezüglich unauffällig ist, wird es sich auch bei den genetischen Mutationen des Kindes um eine “cis-Stellung” handeln, wodurch es lediglich MCAD-Carrier sein kann.
Der “Founder-Effekt” am Beispiel der K329E-Mutation
Die Häufigkeit, mit der speziell die K329E-Mutation weltweit auftritt, verteilt sich sehr unterschiedlich. Während sie in den nordwestlichen Bereichen Europas (durchschnittlich 1:10000 für die homozygote Variante, Carrier ist etwa jede 50. Person), in den USA und besonders in Großbritannien (1:6400, etwa jeder 40. ist Carrier) sehr häufig zu finden ist, zeigt sie sich in den südeuropäischen und afrikanischen Ländern nur sehr selten bis gar nicht. Aufgrund dieser Verteilung nimmt man an, dass speziell diese Mutation von einem einzigen Mitglied eines früheren germanischen Volkes stammt und von diesem an seine Nachkommen vererbt wurde. Somit wird diese Mutation, und damit verbunden der MCAD-Mangel, auch in all den Gegenden besonders häufig angetroffen, in denen sich die Nachfahren dieses ersten “Founders” niedergelassen haben.
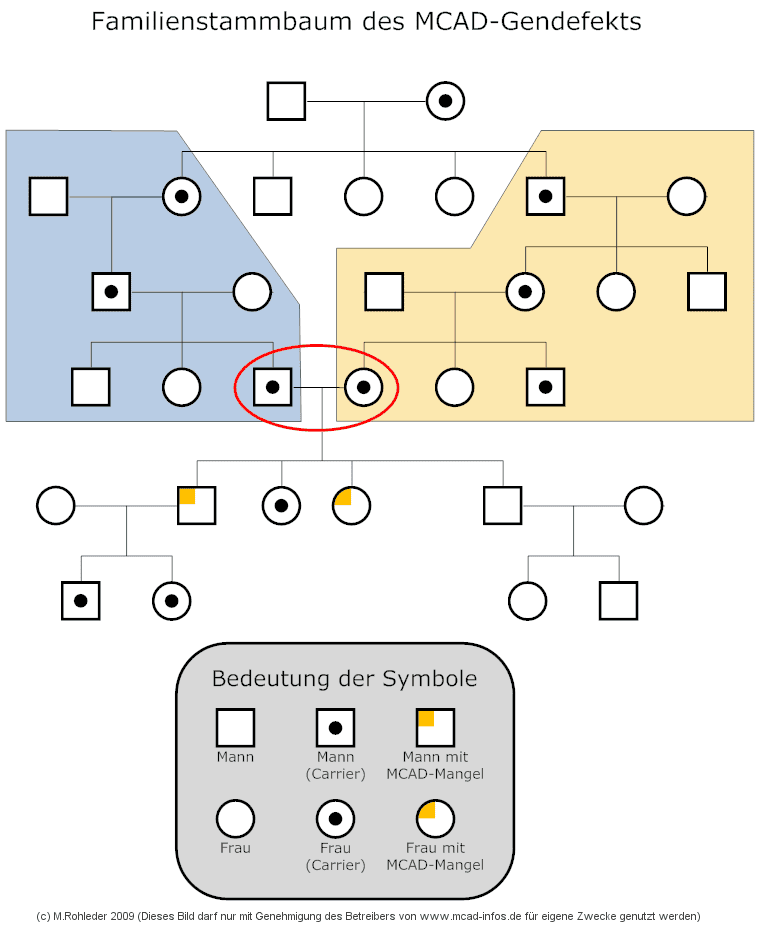
Zur Erklärung des Stammbaumes: Der Founder-Theorie zufolge begann es vor sehr, sehr langer Zeit mit einem Paar, von dem ein Partner als erster Mensch der Welt die K329E-Mutation aufgrund einer spontanen Mutation auf einer Genkopie aufwies und an einige seiner Kinder als Carrier weitergab. Entweder erbten bereits bei diesem Paar, oder bei einem ihrer Nachkommen, gleich zwei oder mehr Kinder den Gendefekt. Aus diesen Kindern entstanden jeweils sich völlig getrennt voneinander entwickelnde Familien (blauer und gelber Bereich). Aufgrund der in früheren Zeiten noch üblichen sehr großen Anzahl von Kindern, gab es neben vielen nicht betroffenen Nachkommen aber auch immer wieder Kinder, die den heterozygoten Gendefekt von einem ihrer Elternteile vererbt bekamen. Alle entstandenen Familien bewirkten eine weitere Verzweigung des Stammbaums. Bei vielen Familienzweigen ergab sich über die Generationen hinweg immer irgendein Vererbungspfad, auf dem die K329E-Mutation erhalten blieb. In irgendeiner Generation ergab es sich dann, dass zufällig ein Carrier-Mitglied der blauen Familie mit einem Carrier-Mitglied der gelben Familie Kinder zeugte (oben mit dem roten Kreis markiert). Aus dieser Verbindung gingen die ersten Kinder mit echtem MCAD-Mangel hervor. Deren Kinder waren auf jeden Fall wieder Carrier und in irgendeiner der folgenden Generationen wiederholte sich dieser Vorgang möglicherweise erneut. Somit kann man schlussfolgern, dass sich die Stammbäume aller Menschen, welche die K329E-Mutation in homozygoter oder heterozygoter Form in sich tragen, zu einem mehr oder weniger weit zurückliegendem Zeitpunkt in einem gemeinsamen Vorfahren treffen müssen.
In gleicher Weise entstanden auch alle anderen Mutationen des ACADM-Gens, so dass inzwischen auch häufig Mischformen, die compound-heterozygoten MCAD-Mangel-Varianten gefunden werden. Manche Mutationen werden im Laufe der Zeit aufgetreten und auch wieder verschwunden sein, da sie nur an sehr wenige Nachkommen durchgereicht wurden. Je häufiger und verbreiteter eine einzelne Mutation inzwischen nachgewiesen werden kann, desto früher muss sie in der Menschheitsgeschichte erstmals aufgetreten sein. Der Ursprung der K329E-Mutation liegt daher vermutlich weit über 2000 Jahre zurück. Andere, seltene Mutationen könnten dagegen auch erst vor wenigen Generationen neu hinzugekommen sein.
Was haben die Kaukasier mit dem MCAD-Mangel zu tun?
Im Zusammenhang mit der Verteilung und Carrier-Häufigkeit des MCAD-Mangels, insbesondere der K329E-Mutation, wird vor allem in englischen MCAD-Abhandlungen häufig betont, dass diese Mutation hauptsächlich bei Europäern (“European Caucasians”) und Nordamerikanern kaukasischer Abstammung auftritt. Dazu muss man aber wissen, dass es dabei nicht speziell um aus dem Kaukasus stammende Menschen geht, sondern dass dieser etwa um 1800 von Johann Friedrich Blumenbach geprägte Begriff auf die von ihm und anderen Anthropologen dieser Zeit aufgestellte Theorie zurückgeht, nach welcher sie versuchten − basierend auf verschiedenen Merkmalen, z.B. Schädelform und Hautfarbe − alle Völker in unterschiedliche “Menschenrassen” zu unterteilen. Diese Rassentheorie wurde seitdem vielfach kritisert und gilt heute als völlig veraltet, jedoch wird der damals geprägte Begriff “kaukasischer Typ” im englischen Sprachraum heute noch als Synonym für grundsätzlich alle europäischen, nordamerikanischen und sonstigen hellhäutigen Menschen gebraucht, da man Anfang des 19. Jahrhunderts den Ursprung all dieser Völker noch im kaukasischen Raum vermutete. Die sehr zweifelhafte Rassentheorie ist Geschichte, der Begriff “kaukasischer Typ” für alle hellhäutigen Europäer und Nordamerikaner hat sich im englischen Sprachgebrauch jedoch gehalten. Aus heutiger Sicht aber hat der MCAD-Mangel also überhaupt nichts mit der Bevölkerung des Gebirges zwischen dem Schwarzen Meer und dem Kaspischen Meer zu tun − auch wenn sich die Behauptung, der MCAD-Mangel beträfe vor allem Menschen kaukasischer Abstammung, inzwischen auch in deutschsprachigen Artikeln vermehrt wiederfindet. Deren Quellen bestehen natürlich hauptsächlich in englischer Literatur, und die deutschen Autoren, die diese Aussagen übernommen haben, wissen vermutlich selbst nichts von der veralteten Rassentherorie des frühen 19. Jahrhunderts als Ursprung dieses Begriffs.
Wo überall auf der Welt gibt es MCAD-Betroffene?
Wenn du das hier liest, weißt du schon, dass du mit dem MCAD-Mangel nicht alleine auf der Welt bist, wie es manchen Eltern in der ersten Zeit nach der Mitteilung des Verdachts bzgl. dieser anfangs noch unbekannten Stoffwechselstörung scheint. MCAD-Carrier und somit auch Kinder und Erwachsene mit MCAD-Mangel gibt es überall auf der Welt, auch wenn ihre Häufigkeit auf einigen Kontinenten sehr viel geringer ist, als hier in Europa, oder in den USA. Verbreitet ist der MCAD-Mangel z.B. in Deutschland, Schweiz, Österreich, Niederlande, Belgien, Dänemark, Frankreich, Italien, Finnland, Griechenland, Ungarn, Großbrittannien, Spanien, Türkei, Island und allen weiteren europäischen Ländern von Russland bis Bulgarien, auch wenn bei den östlicher gelegenen Ländern teilweise andere Genmutationen als die häufige K329E gefunden werden. In allen US-Staaten finden sich Kinder mit MCAD-Mangel, ebenso in Kanada, Argentinien, Australien… In asiatischen Ländern ist der MCAD-Mangel dagegen fast unbekannt − zumindest wird die in den westlichen Ländern am häufigsten vorhandene Mutation K329E unter der asiatischen Bevölkerung so gut wie gar nicht gefunden, falls jemand nicht gerade einen europäischen Vorfahren in seinem Stammbaum hat. In Japan wird der von anderen Mutationen ausgelöste MCAD-Mangel mit einer Häufigkeit von weniger als 1:50000 sogar noch relativ häufig im Screening aufgespürt, so dass sich unter rund 920.000 Geburten pro Jahr (Stand 2018) damit weniger als 20 betroffene Kinder finden.
In manchen der zuvor aufgezählten Länder gibt es nur sehr wenige bekannte Fälle. Allerdings muss dabei immer berücksichtigt werden, dass es in den meisten Ländern der Welt immer noch kein Neugeborenenscreening, geschweige denn ein erweitertes Neugeborenenscreening auf MCAD gibt. Die wenigen bekannten Fälle in diesen Ländern werden daher oft nur aufgrund einer vorangegangenen Entgleisung gefunden, und viele in Wirklichkeit auf den MCAD-Mangel zurückzuführende Todesfälle werden dort vermutlich nach wie vor fälschlicherweise mit dem Reye-Syndrom erklärt oder einfach nur als plötzlicher Kindstod (SIDS) diagnostiziert.
Schweregrade von Mutationen
Der Schweregrad einer ACADM-Mutation lässt sich nur grob einschätzen. Manche missense-Mutationen, von denen jeder Mensch vermutlich gleich ein paar in den verschiedensten Genen − auch dem ACADM-Gen − aufweist, sind so “gut” positioniert, dass sie zu überhaupt keiner feststellbaren Funktionseinschränkung des Enzyms führen (Polymorphismen, s.o.). Allerdings werden solche Kinder dann auch nicht im Screening gefunden. Andere Mutationen wiederum führen zu einem mehr oder weniger reduzierten Restnutzen des Gens. Während und nach der Proteinsynthese faltet sich das erzeugte Protein normalerweise an bestimmten Positionen und bildet eine genau definierte dreidimensionale Molekülstruktur, um dadurch als Enzym wirken zu können .
Einzelne missense-Mutationen können nun dazu führen, dass sich das Molekül an einer falschen Stelle oder im falschen Winkel faltet. Das Resultat ist dann ein nicht voll funktionsfähiges oder instabiles Enzym, dessen Struktur möglicherweise im Zuge eines Infekts mit erhöhter Körpertemperatur kollabiert. Hierzu ist allerdings anzumerken, dass dies eine weitgehend hypothetische Annahme ist, da ein Strukturzerfall mit einhergehendem Aktivitätsverlust des Enzyms nur im Laborversuch mit deutlich über normalem Fieber liegenden Temperaturen festgestellt werden konnte. Die Beobachtungen zeigten, dass ein MCAD-Enzym mit der im weiteren noch beschriebenen häufigsten K329E-Mutation dabei erst bei einer Temperatur von etwa 52°C seine verbleibende Aktivität zur Hälfte eingebüßt hat. Bei Vorliegen einer anderen selteneren Mutation (T168A) trat ein entsprechender Strukturzerfall aber bereits bei 41°C auf und nach ca 20 Minuten Inkubationszeit bei dieser Temperatur hatte das MCAD-Enzym seine Aktivität vollständig eingebüßt. Während eines normalen fieberhaften Infekts werden diese Körpertemperaturen aber nicht erreicht, so dass ein aufgrund der erhöhten Körpertemperatur eintretender Strukturzerfall des Enzyms als Auslöser einer Stoffwechselentgleisung weitgehend ausscheidet. Problematisch ist dagegen oft die den Infekt begleitende Appetitlosigkeit bei gleichzeitig erhöhtem Energiebedarf des Körpers.
Ausser den üblichen ACADM-Mutationen, die zu einer reduzierten MCAD-Aktivität führen, gibt es auch solche, die den Wirkungsbereich des Enzyms verändern. In diesem Bericht aus dem Jahr 1999 werden zwei nahezu identische ACADM-Mutationen verglichen. Die erste führt zu einer reduzierten Enzymaktivität, die sich jedoch über den gleichen Wirkungsbereich wie die Wildtyp-Variante erstreckt. Die zweite Mutation, die an der gleichen Genposition ansetzt und sich nur in Bezug auf den erfolgten Basenaustausch unterscheidet, weist ebenfalls eine deutlich reduzierte Restaktivität auf, aber verschiebt darüber hinaus den Wirkungsbereich des entstehenden Enzyms vom mittelkettigen in den langkettigen Bereich, so dass seine größte Aktivität nicht bei C8, sondern bei C14 und C16 auftritt. Ein Restnutzen bei C8 ist somit fast nicht mehr vorhanden.
Nonsense-Mutationen (Stop-Mutationen) führen, wie zuvor schon erwähnt, zu einem vollständigen Abbruch des Proteinaufbaus und die Schwere dieser Mutationen hängt vereinfacht ausgedrückt davon ab, wieviel DNA-Programmcode vor dem Abbruch noch ordnungsgemäß abgearbeitet werden konnte. Eine zu Beginn der Aminosäuren-Sequenz auftretende Stop-Mutation ermöglicht nur die Bildung eines extrem verkürzten Proteins. Befindet sie sich relativ weit hinten im Bereich des ACADM-Gens, kann das MCAD-Enzym aufgrund des fast vollständig gebildeten Proteins eine vergleichsweise hohe Restaktivität aufweisen. Eine absolut verlässliche Aussage über die tatsächlich zu erwartenden Auswirkungen einer solchen Mutation lässt sich aus diesen Erkenntnissen dennoch nicht ableiten.
MCAD-Mangel-Ausprägungen werden ganz grob in drei unterschiedliche Schweregrade unterteilt:
Schweregrad 1: die sogenannten “milden” Varianten. Diese wurden erst nach der Einführung des erweiterten Neugeborenenscreenings gefunden und zeichnen sich durch hohe Residualaktivitäten (Restaktivitäten) der MCAD-Enzyme, vergleichbar denen von reinen Carriern aus. Die im Acylcarnitin-Profil gemessenen Werte sind selbst in Krankheitszeiten und nach längeren Nüchternzeiten nur geringfügig erhöht. Bei den diesem Schweregrad zugeordneten Varianten ist es fraglich, ob es sich dabei überhaupt um einen als solchen zu bezeichnenden MCAD-Mangel handelt. Da der letzte Beweis der Unbedenklichkeit aber schwer erbracht werden kann, werden auch diese Varianten in den meisten Stoffwechselambulanzen als “normaler” MCAD-Mangel behandelt.
Schweregrad 2: die sogenannten “intermediären” Varianten. Die Risikoeinschätzung dieser MCAD-Ausprägungen ist am kompliziertesten, da sich weder hinsichtlich der Screeningwerte, der Acylcarnitin-Profile, der Mutationen, der Residualaktivitäten noch sonstiger Kriterien eine eindeutige Tendenz in die milde oder in die schwere Richtung erkennen lässt. Die biochemischen Parameter, angefangen mit den Screeningwerten, liegen irgendwo in der Mitte zwischen den typischen milden und den typischen schweren Bereichen, die Residualaktivität des Enzyms wird ebenfalls im mittleren Bereich ermittelt. Größtenteils sind neue Mutationen beteiligt, die auch erst mit der Einführung des erweiterten Neugeborenenscreenings gefunden wurden.
Es gibt aber auch vereinzelte Berichte über Kinder mit erfolgter Entgleisung, deren biochemische Werte ansonsten im Vergleich mit den Werten von schweren Varianten deutlich besser waren, so dass sie von den beteiligen Ärzten als anscheinend “milderer”, also nicht ganz so schwerer MCAD-Mangel eingestuft werden. Das ist aber eine hauptsächlich für die Ärzte interessante medizinische Spitzfindigkeit. Ganz allgemein kann man einen “intermediären” MCAD-Mangel in der Form beschreiben, dass es sich um keine der bekannten schweren Varianten handelt, jedoch die in allen Aspekten im mittleren Bereich liegenden Parameter ein eventuelles Risiko unter ungünstigen Umständen trotzdem als wahrscheinlich erscheinen lassen.
Schweregrad 3: die bekannten Risikovarianten. Hierzu zählt vor allem der klassische MCAD-Mangel (K329E homozygot), sowie eine Reihe weiterer Mutationskombinationen, die ebenfalls in der Vergangenheit mit Entgleisungsfällen dokumentiert wurden. Die beteiligten Mutationen führen zu gravierenden Schäden des MCAD-Enzyms, was sich auch durch extrem niedrige, quasi nicht vorhandene Residualaktivitäten bestätigt.
Vereinfacht ausgedrückt lässt es sich in folgender Form auf den Punkt bringen: Grad 1 − man weiß es nicht, geht aber ziemlich sicher davon aus, dass keine Gefährdung besteht; Grad 2 − man weiß es nicht, geht aber davon aus, dass eine Gefährdung bestehen könnte; Grad 3 − man weiß, dass eine Gefährdung besteht, da die beteiligten Mutationskombinationen bereits zu Entgleisungsfällen geführt haben.
Was ist eine Pseudohomozygotie?
In seltenen Fällen wird bei einem Kind scheinbar eine vorliegende Homozygotie für eine genetische Mutation gefunden, obwohl bei der Untersuchung seiner Eltern nur ein Elternteil diese Mutation aufweist, der andere Elternteil jedoch nicht. In solchen Fällen kann es sich um eine sogenannte Pseudohomozygotie handeln. Wie kommt diese zustande, bzw. wieso wird eine Mutation anscheinende homozygot gefunden, obwohl sie doch nur auf einer Genkopie vorliegt?
Genetische Mutationen werden z.B. dadurch gefunden, dass die in der untersuchten Probe enthaltenen DNA-Stränge, bzw. die darin enthaltenen zu überprüfenden Bereiche, mittels des Verfahrens der Polymerase Chain Reaction (PCR) millionenfach vervielfältigt und danach in kurze Abschnitte zerstückelt werden. Danach kann − ganz vereinfacht ausgedrückt − mit der Waage bestimmt werden, in welcher Menge jeder dieser DNA-Abschnitte vorliegt. Auf der väterlichen und der mütterlichen Genkopie in gleicher Weise existierende Abschnitte werden dabei in “normaler” Menge gefunden. Dagegen findet man von Abschnitten, die sich in der mütterlichen und väterlichen Kopie unterscheiden, jeweils nur die halbe Menge. Wird bei einer solchen Messung eine Mutation nur in halber Menge gefunden (im Vergleich zu den Mengen der um den betreffenden Bereich herum liegenden Abschnitte), dann kann man davon ausgehen, dass diese Mutation nur heterozygot vorliegt. Wird sie aber in der gleichen Menge gefunden, wie die sie umgebenden und mit in die Analyse einbezogenen Abschnitte des DNA-Strangs, dann muss man davon ausgehen, dass diese Mutation auch auf beiden Genkopien existiert und somit homozygot vorliegt.
Es gibt allerdings auch genetische Spielweisen, in denen eine Mutation nur anscheinend in normaler Menge, also vermeintlich homozygot, vorliegt, in Wirklichkeit aber doch nur auf einer Genkopie existiert.
Eine dieser Spielweisen ist eine Sonderform der zuvor schon erwähnten cis-Stellung. Diese liegt vor, wenn durch einen genetischen Defekt ein ganzer Abschnitt eines Allels innerhalb eines Chromosoms verdoppelt wurde. Handelte es sich dabei ausgerechnet um den die Mutation enthaltenden Abschnitt, liegt diese Mutation zwar nur einseitig, aber nun ebenfalls doppelt vor und könnte bei einer molekulargenetischen Untersuchung daher auch nicht nur in halber, sondern in normaler Menge gefunden werden, was dann zunächst zur Annahme einer Homozygotie führen würde. Klarheit kann dann nur die genetische Untersuchung der Eltern schaffen. Findet man bei einem Elternteil ebenfalls beide Mutationen anscheinend homozygot, beim anderen Elternteil dagegen nichts, ist bewiesen, dass es sich um eine Pseudohomozygotie handelt. Der betreffende Elternteil und das Kind sind dann nur Carrier. Eine solche Doppelmutation kann wieder weiter vererbt werden, so dass es in der Bevölkerung neben den “normalen” Versionen des Gendefekts auch die Doppelvariante geben kann, ohne dass es sich dabei um eine die betreffende Stoffwechselstörung auslösende Homozygotie handelt.
Kommt für ein Kind im Neugeborenenscreening aber aufgrund deutlich erhöhter Acylcarnitinwerte ein MCAD-Verdacht auf, ist es nahezu ausgeschlossen, dass es sich bei einer danach homozygot gefundenen Mutation um eine der ohnehin extrem seltenen Pseudohomozygotien aufgrund einer Verdoppelung eines DNA-Abschnitts handeln könnte. Lediglich bei nur minimal erhöhten Acylcarnitinwerten, die von Beginn an eher auf einen Carrierstatus, als auf einen MCAD-Mangel hindeuteten, macht es Sinn, nach einer wider Erwarten gefundenen Homozygotie auch die Eltern hinsichtlich einer möglicherweise vorliegenden Pseudohomozygotie zu untersuchen. Passen dagegen alle vorliegenden Screeningwerte, Acylcarnitinprofile, Sequenzierungsergebnisse und sonstigen Beobachtungen ins Bild eines eindeutigen MCAD-Mangels, ist die Überprüfung einer Pseudohomozygotie unnötig. Ob es im Zusammenhang mit dem MCAD-Mangel überhaupt schon mal einen solchen Fall gegeben hat, ist zweifelhaft − zumindest nicht bezüglich der klassischen K329E-Variante.
Manchmal liegt ein mutierter Abschnitt des DNA-Strangs aber auch deshalb in normaler Menge und somit anscheinend homozygot vor, weil der gesamte vervielfältigte und zerstückelte Bereich der DNA nur von einer einzigen Genkopie (Vater ODER Mutter) herrührt, und es kein entsprechendes Vergleichsmaterial von der Genkopie des anderen Elternteils gibt. In diesem Fall handelt es sich um eine große Deletion, also Lücke auf der zweiten Genkopie, so dass alle hinsichtlich ihrer Menge gemessenen DNA-Stückchen in nur halber Menge vorliegen, dies aber als normale Menge erscheint, und die Mutation somit als Homozygotie angenommen wird. In einem solchen Fall handelt es sich zwar nicht um eine echte Homozygotie, aber trotzdem um einen echten MCAD-Mangel, da auch die zweite Genkopie aufgrund ihrer großen Deletion keine funktionsfähigen Enzyme produzieren kann.
Warum manchmal schon eine einzige Mutation auszureichen scheint
Der Umstand, dass in manchen Gensequenzierungen nur eine einzige Mutation (z.B. eine einzelne K329E) gefunden wird, obwohl die biochemischen Werte des Kindes deutlich auf einen MCAD-Mangel hinweisen, scheint im krassen Widerspruch zu der Aussage zu stehen, dass der Carrierstatus medizinisch ohne Relevanz ist, und ein solches Kind keinen MCAD-Mangel hat.
Um diesen scheinbaren Widerspruch auflösen zu können, muss man sich etwas näher mit den verschiedenen Methoden zur Analyse eines Gendefekts befassen. Es ist nun mal nicht so, dass man sich die im Erbgut eines Kindes enthaltenen, von Vater und Mutter geerbten Genkopien einfach mal unter dem Mikroskop Stück für Stück anschaut und dann sofort sieht, wo es Abweichungen vom Wildtyp gibt. Zur Analyse bedient man sich einer ganzen Reihe hochkomplizierter chemischer Verfahren, die jedes für sich genommen, für eine ganz bestimmte Analyseaufgabe entwickelt wurden. Je nach angewendetem Verfahren können ganz bestimmte Arten von Gendefekten gefunden werden, andere aber nicht.
Die üblicherweise zur Mutationsbestimmung durchgeführte Form der Gensequenzierung kann z.B. keine Deletions detektieren. Deletions sind, wie oben beschrieben, kleinere oder größere Lücken innerhalb des DNA-Strangs, so dass im fortlaufenden Code eine einzelne oder auch mehrere zusammenhängende Basen fehlen. Es sind Fälle bekannt, in denen sich eine Deletion über mehrere Exons ausgedehnt hat. In der Gensequenzierung fand man zwar die häufige K329E-Mutation, jedoch nur heterozygot, denn die andere Genkopie wies an der Stelle die Wildtyp-Basensequenz auf. Die Untersuchung der anderen Exons brachte keine weiteren abweichenden Basensequenzen zutage. Es konnte mithilfe der Gensequenzierung jedoch nicht gezeigt werden, dass in der vom anderen Elternteil vererbten Genkopie eine riesige Lücke vom ersten bis zum sechsten Exon vorlag. Letztendlich kann die Gensequenzierung nur das sequenzieren, was auch da ist. Lücken auf einer Genkopie können nicht sequenziert werden, und daher lassen sich dann an diesen Stellen auch keine Abweichungen zur anderen Genkopie feststellen.
Nicht immer sind die Deletions so groß, aber auch kleinere Deletions werden meist erst dann gefunden, wenn man aufgrund des nicht zufriedenstellenden Ergebnisses der Gensequenzierung noch andere Verfahren, z.B. die aCGR (array-based Comparative Genomic Hybridization) anwendet. Dieses Verfahren taugt nun wiederum überhaupt nicht zur Feststellung und Benennung einzelner Mutationen, kann aber auf einer Art DNA-Zählbrett im Vergleich mit einer Referenz-DNA sehr genau aufzeigen, in welchen Bereichen die zu untersuchende DNA Deletions aufweist. Neuere Untersuchungen legen die Vermutung nah, dass es sich bei den manchmal mit der Gensequenzierung nicht auffindbaren zweiten Mutationen häufig um kleine oder größere Deletions handelt, denen man mit anderen Verfahren nachspüren müsste.
Solche weiterführenden Untersuchungen werden aber in den seltensten Fällen durchgeführt, denn sie sind ebenfalls sehr aufwändig, zeitintensiv und daher auch teuer. Für die MCAD-Diagnose reicht es den Uni-Kliniken in den meisten dieser ohnehin nur seltenen Fälle, wenn die biochemischen Werte einen MCAD-Mangel vermuten lassen, und dies durch die Auffindung wenigstens einer bekannten MCAD-Mutation unterstützt wird. Es muss dann zwar zwingend noch ein Defekt an der vom zweiten Elternteil beigesteuerten Genkopie existieren, aber die Suche danach wird in den meisten Fällen als nicht mehr notwendig angesehen. Überhaupt wird in den letzten Jahren vor allem die heutzutage sehr genaue und verlässliche Residualaktivitäts-Messung als Standardverfahren zum sicheren Nachweis des MCAD-Mangels durchgeführt und die früher übliche und sehr teure Suche nach den beteiligten Mutationen ist auf dem Rückschritt. Letztlich ist es ja auch völlig belanglos, wie die bei dem Kind vorliegenden Mutationen nun genau heißen. Von Bedeutung ist dagegen, welche Aktivität die von dem Kind gebildeten MCAD-Enzyme noch entfalten können, wenn es darauf ankommt, bzw. welchem Schweregrad die MCAD-Mangel-Ausprägung zugeordnet werden kann.
Milde Verlaufsformen
Schon Mitte der 90er Jahren haben entsprechende Forschungen gezeigt, dass auch im compound-heterozygoten Fall, also beim Vorliegen zweier unterschiedlich mutierter Allele, das Prinzip “Funktion dominiert über Funktionsmangel” gilt. Die Mutation A mit der höheren Restleistung dominiert über die schwerwiegendere Mutation B, so wie das Wildtyp-Allel im rein heterozygoten Fall grundsätzlich über das mutierte Allel dominiert. Selbst wenn die schwerere Mutation B zu einer kompletten Funktionseinbuße des entstehenden Enzyms führen würde, wirkt sie sich in so einem Fall nicht aus, und die resultierende Form des MCAD-Mangels wird bei dem Patienten nur durch die mildere Mutation A bestimmt. In den Untersuchungen der vergangenen Jahre wurde deshalb festgestellt, dass bestimmte Mutationen eine harmlosere Ausprägung eines MCAD-Mangels erwarten lassen, als beispielsweise die klassische Variante K329E homozygot. Medizinisch wird dieser Zustand als “Non-Disease” (Nicht-Krankheit) bezeichnet. Fest machte man diese Vermutung vor allem daran, dass die den auffälligen Neugeborenen-Screenings folgenden Genanalysen eine ganze Reihe Mutationen des ACADM-Gens aufgezeigt haben, die bislang nicht in Form einer Stoffwechselentgleisung in Erscheinung getreten sind, obwohl bei den gefundenen Mutationen oft K329E compound heterozygot beteiligt ist.
Zum Beispiel verursachen die Mutationen Y67H compound heterozygot mit K329E, sowie die Mutationen G267R homozygot, S245L homozygot und weitere seltene Mutationen eine vermutlich milde (genauer: benigne = gutartige) Variante des MCAD-Mangels [Quelle]. Speziell bei den beiden zuletzt genannten Mutationen ist diese Einschätzung aber mit äusserster Vorsicht zu genießen, da diese Annahmen in beiden Fällen auf bisher jeweils lediglich einem einzigen untersuchten Fall eines Kindes mit zudem noch blutsverwandten Eltern zu beruhen scheinen. In compound heterozygoter Form ist zumindest die G267R in der weiter zurückliegenden Vergangenheit bereits mehrfach im Rahmen schwerer Entgleisungen zu Tage getreten. Daher ist äußerst fraglich, warum sie z.B. in Kombination mit einer bekanntermaßen schweren Mutation wie K329E einen milden MCAD-Mangel bewirken sollte.
Im Allgemeinen ist eine milde Form dadurch charakterisiert, dass zwar eine für den MCAD-Mangel typische deutliche Verringerung der Aktivität des MCAD-Enzyms messbar ist (unter 50% der Aktivität des Enzyms bei Personen ohne MCAD-Mangel), aber die im Screening ermittelten Werte C8:0 (Octanoylcarnitin) und C8:0/C10:0 (Verhältnis von Octanoylcarnitin zu Decanoylcarnitin), sowie weitere Sekundärparameter nur verhältnismäßig leicht über dem zugrunde gelegten Normbereich liegen. Besonders das Verhältnis von C8:0 zu C10:0 ist beim klassischen MCAD-Mangel normalerweise sehr groß (in einer niederländischen Studie ermittelte man dafür Werte zwischen 8,4 und 18,4, während die milden Ausprägungen nur zwischen 1,4 und 2,3 lagen).
Das medizinische Dilemma mit den milden Varianten
Das erweiterte Neugeborenenscreening auf eine Reihe zusätzlicher Stoffwechselstörungen ist genaugenommen nichts weiter als der Versuch einer frühestmöglichen Vorhersage über möglicherweise im weiteren Leben auftretende gesundheitliche Probleme. Ob diese vermuteten gesundheitlichen Probleme unter ungünstigen Umständen, wie z.B. langen Nahrungspausen, auch wirklich eintreten würden, ist nicht im Geringsten sicher. Aus den bisher untersuchten Fällen kann man nur eine Reihe von Indizien dafür ableiten, wie wahrscheinlich es ist, dass bei dem Neugeborenen ein behandlungsbedürftiger MCAD-Mangel vorliegt. Besonders beim Auffinden der als riskant bekannten Mutationskombinationen ist aufgrund der bisherigen Erfahrungen davon auszugehen, dass es ohne diese frühzeitige Erkennung und entsprechend ausgerichtete Behandlung, mit sehr hoher Wahrscheinlichkeit früher oder später zu einer Krankheitssituation mit einhergehender Stoffwechselentgleisung kommen würde. Die frühe Diagnose kann bezogen auf diese Kinder also durchaus als lebensrettend angesehen werden. Dem gegenüber steht die große Zahl der aufgrund der notwendigerweise sehr niedrigen Schwellwerte gefundenen, vermutlich milden Varianten. Diese stellen zwar sowohl genetisch, als auch enzymatisch betrachtet unbestreitbar einen “echten”, d.h. im Labor biochemisch nachweisbaren MCAD-Mangel dar, in ihrer realen Ausprägung führen sie jedoch fast immer zu keinerlei spürbaren Einschränkungen im Leben eines betroffenen Menschen. Den Ärzten ist bewusst, dass die Mitteilung einer solchen, lediglich auf Laborwerten basierenden Diagnose, für das Kind und seine Familie mehr Nach- als Vorteile bringen kann, da es höchstwahrscheinlich selbst ohne die nun mal bereits erfolgte Frühdiagnose niemals in die Gefahr einer Entgleisung geraten würde. Wurde die Mitteilung über den vermutlich vorliegenden MCAD-Mangel jedoch erst einmal ausgesprochen, werden sowohl das Kind, als auch seine Eltern ihr Leben lang von dieser ständig über ihnen schwebenden Ahnung begleitet, die sich in verschiedenen Situationen zu einer starken psychischen Belastung ausweiten kann. Im Gegensatz dazu gibt es höchstwahrscheinlich unzählige Menschen in Deutschland und auf der Welt, die hervorragend mit ihrem milden MCAD-Mangel leben − ohne davon zu wissen und ohne sich die geringsten Gedanken darüber zu machen. Um dies anhand von ein paar angenommenen Zahlen zu verdeutlichen:
In Deutschland dürfte es aufgrund der derzeit angenommen Häufigkeit des MCAD-Mangels quer durch alle Altersgruppen zwischen 5500 und 8000 Menschen geben, die von dieser Stoffwechselstörung in ihrer milden oder normalen Ausprägung betroffen sind. Da das erweiterte Screening aber erst seit wenigen Jahren durchgeführt wird, wissen davon aber hauptsächlich nur diejenigen Kinder, die ab dem Jahr 2000 geboren wurden, und selbst damals wurde nur im Rahmen von Modellversuchen das Screening auf den MCAD-Mangel ausgedehnt. Bei einer durchschnittlichen Geburtenrate von rund 700.000 Kindern pro Jahr in ganz Deutschland, und somit zwischen 50 und 70 neuen MCAD-Fällen pro Jahr, wissen bis zum heutigen Tag (2020) also gerade mal 1000 bis 1400 Menschen, dass sie vom MCAD-Mangel (in milder oder normaler Ausprägung) betroffen sind. Unter den vor 2000 geborenen Deutschen wissen also − mit wenigen Ausnahmen − 4000 bis 6000 Personen gar nicht, an was sie im medizinischen Sinne eigentlich “leiden”. Da man davon ausgehen muss, dass rund die Hälfte aller mit aktuellen Methoden aufgespürten Betroffenen die Risikovariante des MCAD-Mangels aufweist, wird es unter diesen früheren Jahrgängen viele Betroffene gegeben haben, die tatsächlich bereits in frühester Kindheit in Folge einer nicht als solche erkannten Stoffwechselkrise verstorben sind. Trotzdem bleiben immer noch mindestens 2000-3000 Personen mit unerkanntem milden MCAD-Mangel, die auch ohne dieses Wissen schon als Kleinkind sehr gut lebten und sich nicht die geringsten Gedanken darüber machen, wie lange sie nachts schlafen, was sie essen oder wie lange die Pausen zwischen ihren Mahlzeiten sein dürfen.
Verschiedene Länder praktizieren hinsichtlich dieses Dilemmas unterschiedliche Herangehensweisen [Zschocke 2008, “Dominant vs. recessive: Molecular mechanisms in metabolic disease”]. In manchen Ländern werden die Ergebnisse des Neugeborenen-Screenings sehr viel zurückhaltender behandelt und als mild angenommene Varianten den Eltern gegenüber auch deutlich weniger dramatisiert mitgeteilt. In anderen Länder herrscht eine generell größere Vorsicht, so dass dort annähernd alle im Screening aufgefundenen Varianten des MCAD-Mangels von Anfang an als gleichermaßen kritisch angesehen und entsprechend konsequent behandelt werden.
Wie sich eine milde Form im Leben eines davon betroffenen Menschen genau äussert, und ob das Risiko für eine Stoffwechselentgleisung tatsächlich deutlich geringer ist, als im klassischen Fall, ist zwar anzunehmen, aber noch nicht ausreichend erforscht. Entsprechende Studien werden z.B. in Deutschland nämlich schon alleine dadurch erschwert, dass hier die allgemeinen Empfehlungen genau wie bei den eindeutig riskanten MCAD-Varianten dahin gehen, grundsätzlich größere Fastenperioden zu vermeiden. Momentan ist es aufgrund des medizinischen Kenntnisstandes einfach noch die klügere Entscheidung, sich auch hinsichtlich der milden Varianten auf der sicheren Seite zu bewegen − allerdings kommt man dann mit den diesbezüglichen Erkenntnissen natürlich auch nicht weiter, was letztlich auf Kosten der Kinder und ihrer Familien geht.
Aus dem Genotyp folgt nicht der Phänotyp!
Würde man die Gesamtheit der Gene eines einzelnen Lebewesens entschlüsseln, könnte man daraus theoretisch eine Liste der Merkmale erstellen, die bei ihm gemäß genetischer Programmierung vorliegen müssen. Die Gesamtheit dieser durch die Gene vorgegebenen Eigenschaften bezeichnet man als “Genotyp”. Wie sich diese Eigenschaften bei dem Lebewesen aber tatsächlich ausprägen, wird durch alle möglichen Umwelteinflüsse während seiner Entwicklung beeinflusst.
Mit Umwelteinflüssen sind dabei nicht nur klimatische Bedingungen gemeint, sondern der gesamte Lebensraum, bzw. die Lebensumstände des betreffenden Individuums. Natürlich wird es sich auf einen Menschen unterschiedlich auswirken, ob er am Nordpol, am Äquator oder mittendrin wohnt; ob er sich hauptsächlich von Fisch, Fleisch oder Gemüse ernährt; ob er sich täglich an einen reich gedeckten Tisch setzen kann oder ob er sich sein Essen mit großer körperlicher Anstrengung täglich selbst jagen oder gar mühsam aus unfassbar großen, giftige Dämpfe ausstoßenden Müllbergen zusammensuchen muss; ob er bei einer Erkrankung Medikament A, Medikament B oder gar keins bekommt. Es sind unzählige Einflüsse, die sich alle in irgendeiner Weise auf den Menschen als komplexen Organismus auswirken.
Diese Umwelteinflüsse sind aber nicht grundsätzlich negativ zu werten. Manche haben sicherlich schädliche Folgen, andere wiederum positive Auswirkungen. Wieder andere können weitgehend neutral bewertet werden. Die Gesamtheit aller dieser Einflüsse, die von Zeitpunkt der Zeugung an zeitlebens auf den Menschen einwirken, führen zu seiner ganz individuellen Ausprägung der genetisch programmierten Merkmale. Diese individuelle Ausprägung bezeichnet man als “Phänotyp”. Zwar haben eineiige Zwillinge zeitlebens den gleichen Genotyp, da bei ihnen alle genetischen Eigenschaften identisch sind, im Lauf der Zeit kann sich ihr Phänotyp aber deutlich unterscheiden − besonders dann, wenn sie in Umgebungen mit sehr unterschiedlichen Umweltbedingungen leben, bzw. aufwachsen. Sie sind zwar nach wie vor deutlich als Zwillinge erkennbar, lassen sich aber anhand vieler individueller Merkmalsausprägungen unterscheiden. Man kann also von einem Genotyp ausgehend lediglich vage Vermutungen über den dazugehörigen Phänotyp anstellen, ihn aber nicht hundertprozentig genau vorhersagen.
Genau so verhält es sich den wissenschaftlichen Erkenntnissen zufolge auch mit der individuellen Ausprägung des MCAD-Mangels. Würde man mehrere (gleichaltrige) Kinder, die alle den gleichen Gendefekt (z.B. K329E homozygot) haben, über einen längeren Zeitraum hinweg beobachten, könnte man feststellen, dass sich der aus diesem Defekt resultierende MCAD-Mangel bei allen diesen Kindern in unterschiedlicher Weise ausprägt. Ein Kind übersteht Nachtnüchterheitsphasen von 12 Stunden grundsätzlich problemlos, bei einem anderen Kind würde sich schon nach 8 Stunden ein Energiemangel anbahnen. Ein in einer Gegend mit nachweisbar sauberer Luft lebendes Kind hat möglicherweise noch nie auch nur die leichtesten Anzeichen einer Unterzuckerung gezeigt, während ein in der Großstadt aufwachsendes Kind schon mehrfach aufgrund einer simplen Erkältung eine Glucose-Infusion benötigte.
Diese zuletzt genannten Situationen sind aber nur zur Verdeutlichung des Sachverhalts herangezogene Beispiele und keine nachgewiesenen positiven oder negativen Umwelteinflüsse. Fest steht jedoch, dass man genetisch festgelegte Merkmale nicht isoliert betrachten kann, sondern dass jeder Mensch das individuelle Gesamtresultat aus allen seinen Merkmalen und den auf ihn einwirkenden Einflüssen ist, so dass die phänotypische Ausprägung des MCAD-Mangels bei verschiedenen Kindern, selbst bei Geschwistern, trotz diesbezüglich gleichem Genotyp drastisch voneinander abweichen kann und wird. Auch die Ergebnisse regelmäßiger Blut- und Urinuntersuchungen können bei allen diesen Kindern stark unterschiedlich ausfallen. Selbst dann, wenn ein Kind eine lange bekannte und häufige ACADM-Mutation aufweisen sollte, müssen die behandelnden Ärzte diesem Umstand Rechnung tragen und dürfen nicht strikt nach Schema f vorgehen. Jedes Kind mit MCAD-Mangel sollte einer individuell abgestimmten Diagnose und Therapie zugeführt werden. Dies ist jedoch nur unser aller Wunschdenken − in der Realität werden oft alle Kinder nahezu gleich behandelt, egal welche Variante von MCAD-Mangel sie aufweisen und wie stark oder schwach er sich bei ihnen auswirkt.
Ich möchte an dieser Stelle noch einmal ausdrücklich darauf hinweisen, dass ich als Autor dieser Seiten größten Wert auf Korrektheit gelegt habe, aber ansonsten medizinischer Laie bin. Alle veröffentlichten Informationen wurden von mir anhand verschiedener verfügbarer Promotionen, Studienarbeiten, medizinischen Veröffentlichungen, MCAD-Broschüren, Merkblätter, Webseiten von Kliniken, Aussagen der Stoffwechselambulanz und sonstigen Erkenntnissen und Erfahrungen (eigenen und denen unserer Forumsteilnehmer) zusammengetragen und in inzwischen weit über 1000 Stunden Arbeitszeit gelesen, unzählige Male überdacht, sortiert und schließlich in der jetzigen Form hier niedergeschrieben. Das Lesen dieser Seiten darf aber auf keinen Fall den Besuch beim Arzt oder der Stoffwechselambulanz ersetzen, sondern soll lediglich dazu dienen, schon mal etwas besser über die ganze Thematik und Problematik Bescheid zu wissen!